O czym użytkownik chromatografu jonowego powinien zawsze pamiętać
Autor: Rajmund Michalski*
Streszczenie
Chromatografia jonowa jest obecnie najważniejszą instrumentalną metodą oznaczania jonów przede wszystkim w wodach i ściekach. Chromatograf jonowy to wydatek o wartości zazwyczaj kilkuset tysięcy złotych, ale doświadczony analityk obsługujący tak wyrafinowany sprzęt to inwestycja bezcenna.
W dwóch częściach niniejszego artykułu opisano najważniejsze zasady prawidłowej pracy z chromatografem jonowych, takie jak dobór właściwej metody przygotowania próbki do analizy; wybór kolumny i eluentu oraz sposobu detekcji; wpływ temperatury czy stężenia eluentu na jakość otrzymywanych wyników; zasady przechowywania kolumn oraz metody rozwiązywania problemów podczas użytkowania chromatografu jonowego.
Wprowadzenie
Chromatografia jako metoda analityczna została odkryta i opisana w roku 1903 przez rosyjskiego biochemika Michała Tswetta, pracującego na Uniwersytecie Warszawskim. Rozdzielił on barwniki roślinne, wykorzystując do tego celu kolumnę wypełnioną węglanem wapnia. W celu opisania tej metody greckimi słowami „barwa” i „zapis” utworzył nowe słowo „chromatografia” [1]. Przez kolejne kilkadziesiąt lat chromatografia nie znalazła szerszego uznania i dopiero opracowanie przez Martina i Snydera teorii rozdzielania, za co otrzymali Nagrodę Nobla, a na której oparte są wszystkie metody chromatograficzne, zmieniło diametralnie podejście do tej techniki separacyjnej.
Najszybciej rozwinęły się metody chromatografii cienkowarstwowej i gazowej, a nieco później trudniejsza do opracowania pod względem technicznym chromatografia cieczowa, która stosowana była przede wszystkim do rozdzielania i oznaczania związków organicznych.
Jej zastosowanie do analiz substancji nieorganicznych było ograniczone, co wynikało z dwóch zasadniczych powodów. Po pierwsze stosując detektor konduktometryczny, który z oczywistych powodów wydawał się być detektorem „dedykowanym” do analiz jonów, nie było możliwe odróżnianie przewodnictwa jonów analitu od przewodnictwa jonów eluentu. Po drugie istniejące i stosowane z powodzeniem metody oznaczania jonów nieorganicznych, jak metody miareczkowe, wagowe, spektrofotometryczne i elektroanalityczne pomimo pewnych ograniczeń były tanie i łatwo dostępne.
Na przełomie lat 60-tych i 70-tych XX wieku prowadzono intensywne badania nad wykorzystaniem znanego od wielu lat zjawiska wymiany jonowej do chromatograficznego rozdzielania i oznaczania anionów i kationów. Przełom w tych pracach nastąpił w roku 1971 kiedy to Hamish Small i jego współpracownicy pracujący dla firmy Dow Chemicals opracowali i opisali chromatograficzną metodę oznaczania jonów litu, sodu i potasu z wykorzystaniem detekcji konduktometrycznej [2]. Oficjalnie za datę powstania chromatografii jonowej uważa się wrzesień 1975 roku, kiedy to na corocznym zjeździe Amerykańskiego Towarzystwa Chemicznego w Chicago, firma Dionex zaprezentowała pierwszy handlowy chromatograf jonowy, a praca Smalla, Stevena i Baumanna [3] uznana została za kamień milowy w rozwoju tej nowoczesnej metody analitycznej.
Obecnie chromatografia jonowa jest najpopularniejszą instrumentalną metodą oznaczania anionów i kationów stosowaną w większości laboratoriów na całym świecie [4]. Około 90% wszystkich chromatografów jonowych jest wykorzystywanych przede wszystkim do oznaczania głównych nieorganicznych anionów (F–, Cl–, NO2–, NO3–, PO43- i SO42-) oraz kationów (Na+, K+, NH4+, Ca2+ i Mg2+) w wodach i ściekach. Jej najważniejsze zalety to [5]:
- możliwość jednoczesnego oznaczania kilkunastu jonów;
- krótki czas analizy;
- niskie granice wykrywalności;
- niewielka ilość próbki potrzebna do analizy;
- możliwość stosowania różnych detektorów (konduktometryczne, UV/Vis, fluorescencyjne, refraktometryczne, amperometryczne, potencjometryczne, ICP-MS, MS, chemiluminescencyjne);
- prosty sposób przygotowania (w przypadku analizy wód i ścieków zazwyczaj wystarcza przesączenie próbki przez filtr o wymiarach porów 0,45 mm);
- możliwość jednoczesnego oznaczania kationów i anionów, lub jonów organicznych i nieorganicznych;
- wysoka selektywność w stosunku do oznaczanych substancji w próbkach o złożonej matrycy [6];
- stosowanie tanich i bezpiecznych eluentów;
- możliwość oznaczania jonów tego samego pierwiastka na różnych stopniach utlenienia (analityka specjacyjna min. Cl–/ClO2–/ClO3–/ClO4–; Br–/BrO3–; NO2–/NO3–/NH4+; Cr(III)/Cr(VI), Fe(II)/Fe(III), As(III)/As(V), Se(IV)/Se(VI)) [7-9].
Wymienione zalety chromatografii jonowej spowodowały, że znajduje ona szerokie zastosowania min. do: oznaczania anionów i kationów w wodach do spożycia, wodach powierzchniowych, wodach podziemnych, ściekach przemysłowych i komunalnych, a także do badań wybranych substancji min. w: rolnictwie, galwanotechnice, medycynie, czy przemyśle spożywczym [10]. Chromatografia jonowa jest metodą referencyjną oznaczania wybranych nieorganicznych anionów i kationów w wodach, a Międzynarodowa Organizacja Normalizacyjna (ISO) i w ślad za nią Polski Komitet Normalizacyjny opublikowały szereg metod znormalizowanych dotyczących oznaczania jonów w wodach, ściekach i powietrzu [11].
Problemy związane z użytkowaniem chromatografu jonowego dotyczą zazwyczaj niewłaściwego rozdzielania jonów, wysokiego tła eluentu, nieregularnej linii podstawowej, skracania się czasów retencji jonów i zmian charakterystyki kolumny analitycznej. W związku z powyższym należy pamiętać o takich podstawowych czynnościach jak: odpowiednie przygotowanie próbki do analizy (szczególnie w przypadku próbek o obciążonej matrycy), odgazowaniu eluentu; doborze właściwej pod względem pojemności jonowymiennej i selektywności kolumny analitycznej; właściwym wyborze eluentu; a także odpowiednich parametrach pracy supresora i detektora, oraz odpowiednim sposobie przechowywania kolumn [12].
Te zagadnienia, czyli o czym każdy analityk – użytkownik chromatografu jonowego powinien pamiętać opisano w dwóch częściach niniejszego opracowania.
Trudne początki, czyli jak kupić to co naprawdę potrzebujemy
Postępowanie podczas analizy jonów z wykorzystaniem chromatografu jonowego nie różni się zasadniczo od wykonywania analiz innymi metodami instrumentalnymi, a w szczególności chromatograficznymi. Opracowując specyfikację przetargową powinniśmy pamiętać nie tylko o cenie przyrządu (zazwyczaj sprzedawcy wiedzą wcześniej od nas ile możemy wydać), ale także o okresie gwarancyjnym, kosztach bieżącej obsługi czy warunkach serwisowania. Pamiętajmy także o tym, do czego nasz upragniony chromatograf jonowy ma służyć – czyli unikajmy przerostu formy nad treścią. Znanych jest wiele przypadków zakupu super sprzętu za ponad 300 000 zł do najprostszych rutynowych analiz, i odwrotnie sprzętu podstawowego, podczas gdy nasze potrzeby analityczne są znacznie bardziej wyrafinowane. Zakup wysokiej klasy aparatury analitycznej takiej, jak chromatograf jonowy, często nie jest końcem, a ……. początkiem problemów. I to nie dlatego, że sprzęt ten jest zły lub niewłaściwy w stosunku do naszych potrzeb, lecz dlatego, że użytkowania tak wyrafinowanego sprzętu wymaga często dodatkowych akcesoriów oraz czasu ze strony analityka, który musi go „poczuć”. Sentencja „Czas jest ojcem prawdy” jest tutaj jak najbardziej na miejscu.
Na pewno nie jest tak, jak czasami uważają nasi zwierzchnicy, że kupiliśmy „czarną skrzynkę” i ona sama rozwiąże wszystkie nasze problemy analityczne.
Poza zakupem aparatu potrzebne są jeszcze m.in. dodatkowe kolumny, zestawy do przygotowania próbek, odpowiedniej czystości woda, odczynniki chemiczne, wzorce czy materiały referencyjne. W tej sytuacji okazuje się często, że najtańszym elementem tej inwestycji jest … analityk, który musi zapoznać się z działaniem tego skomplikowanego urządzenia, także być gwarantem terminowości, rzetelności, i jakości uzyskiwanych wyników analitycznych. Czy rozsądne jest, że sprzęt wartości kilkuset tysięcy złotych obsługuje osoba zarabiająca ułamek procenta jego wartości? Niestety takie są polskie realia. Tym niemniej trudno się dziwić, że kiedy osoba taka zdobędzie już niezbędne doświadczenie oczekuje czegoś więcej.
Na rynku światowym, jak i polskim w zakresie produkcji i sprzedaży chromatografów jonowych (oraz akcesoriów do nich) wiodącą rolę odgrywają dwie firmy – amerykański Dionex oraz szwajcarski Metrohm. Ciekawostką jest to, że firma Dionex sprzedaje swoje chromatografy jonowe w wersji z supresorami zarówno do oznaczania anionów jak i kationów, natomiast firma Metrohm supresję zaleca i stosuje wyłącznie do oznaczania anionów.
Kiedy już zakupimy wymarzony sprzęt i przebrniemy przez etap instalacji, jak i podstawowej obsługi urządzenia oraz zaznajomimy się z programem, nadchodzi czas przeprowadzenia pierwszych analiz.
Przygotowanie próbek do analizy, czyli to co najważniejsze
Przygotowanie próbek stanowi kluczowy etap analizy i najczęściej to on, a nie inne etapy decydują o jakości uzyskanych wyników. W przypadku analiz próbek silnie zanieczyszczonych należy usunąć nadmiar jonów dominujących (zazwyczaj Cl–, SO42-, Na+) za pomocą specjalnych kolumienek, lub rozcieńczyć próbkę wodą dejonizowaną. W obydwu przypadkach istnieje ryzyko utraty części analitu występujących na niższych poziomach stężeń i popełnienie dużych błędów pomiarowych.
Próbki przed analizą należy przesączyć przez filtr o średnicy porów 0,45 μm. Nowoczesne chromatografy jonowe mają wbudowane odpowiednie zestawy do filtracji. Niemniej w celu wydłużenia żywotności kolumny rozdzielającej wstępne przesączenie próbki jest zalecane. O ile w przypadku oznaczania jonów w wodach i ściekach zazwyczaj wystarcza jej przesączenie przez filtr o wymiarach porów 0,45μm, w przypadku analiz próbek stałych, gazowych czy ciekłych o obciążonej matrycy konieczne jest stosowanie innych bardziej wyrafinowanych metod przygotowania próbek [13].
Wszystkie próbki, w tym próbki wzorcowe, roztwory do regeneracji, oraz woda i eluenty powinny być wolne od cząsteczek stałych, które mogą powodować wzrost ciśnienia wstecznego w kolumnie rozdzielającej i zmieniać jej charakterystykę. Dotyczy to szczególnie eluentu, który stale przepływa przez kolumnę (od kilkuset ml do powyżej 1,5 dm3 w ciągu dnia pracy, podczas gdy objętość próbki jest kilka tysięcy razy mniejsza).
Próbki przeznaczone do oznaczania anionów nie mogą być utrwalane za pomocą kwasów, tak jak jest to zalecane w innych metodach analizy, ponieważ wprowadzalibyśmy wtedy do próbki dodatkowo duże ilości jonów, które mogą przeszkadzać w rozdzielaniu i przeładowywać kolumnę. Ustalając strategię pobierania, przechowywania oraz oznaczania należy pamiętać o nietrwałości wielu jonów. Czas i sposób przechowywania próbek przeznaczonych do oznaczania poszczególnych jonów techniką chromatografii jonowej przedstawiono w tabeli 1.
Tabela 1. Metody konserwacji i czas przechowywania próbek wodnych analizowanych za pomocą chromatografii jonowej
|
Jony analitu |
Zalecana czynność |
Maksymalny czas przechowywania próbki [dni] |
|
Azotany(III) |
Schłodzenie do temp. 4°C |
2 |
|
Azotany(V) |
Schłodzenie do temp. 4°C |
2 |
|
Bromiany(V)* |
Dodanie 50 mg/dm3 etylenodiaminy |
28 |
|
Bromki |
Nie jest wymagana |
28 |
|
Chlorany(III)* |
Dodanie 50 mg/dm3 etylenodiaminy, schłodzenie do temp. 4°C |
14 |
|
Chlorany(V)* |
Dodanie 50 mg/dm3 etylenodiaminy |
28 |
|
Chlorki |
Nie jest wymagana |
28 |
|
Chromiany |
Ustalenie wartości pH próbki do 9-9,5 za pomocą np. NH4OH |
1 |
|
Cyjanki |
Ustalenie wartości pH próbki powyżej 12 za pomocą NaOH, schłodzenie do temp. 4°C |
14 |
|
Fluorki |
Nie jest wymagana |
28 |
|
Octany |
Schłodzenie do temp. 4°C |
2 |
|
Mrówczany |
Schłodzenie do temp. 4°C |
2 |
|
Fosforany(V) |
Schłodzenie do temp. 4°C |
2 |
|
Jony amonowe |
Filtracja, schłodzenie do temp. 4°C |
7 |
|
Jony magnezowe |
Filtracja |
42 |
|
Jony metali (m.in. Co, Ni, Zn) |
Zakwaszenie roztworu do wartości < 2 za pomocą np. st. HNO3 |
6 miesięcy |
|
Jony potasowe |
Filtracja |
42 |
|
Jony sodowe |
Filtracja |
42 |
|
Jony wapniowe |
Filtracja |
42 |
|
Siarczany(VI) |
Nie jest wymagana |
28 |
* Próbki przeznaczone do oznaczania chloranów(III), chloranów(V) i bromianów(V) natychmiast po pobraniu powinny być przepłukane gazem obojętnym (np. helem, argonem lub azotem), a próbki do oznaczania chloranów(III) powinny być przechowywane w pojemnikach z ciemnego szkła [14]
Próbki oraz roztwory do kalibracji nieorganicznych jonów powinny być przechowywane w lodówce w temp. + 4ºC w naczyniach z tworzyw sztucznych. Naczynia szklane nie są odpowiednie ze względu na ryzyko wymywania z nich m.in. jonów z fluorkowych i sodowych. W przypadku oznaczania kationów zaleca się zakwaszenie próbki do pH=2,5 do 3,5 za pomocą np. kwasu azotowego(V), co poprawia powtarzalność uzyskiwanych wyników, szczególnie dla później wymywanych jonów dwuwartościowych.
Woda stosowana w chromatografii jonowej powinna mieć przewodnictwo elektryczne właściwe na poziomie 18 MΩ·m-1, czyli 0,05 μS/cm. Wyższe przewodnictwo wskazuje na obecność w niej pewnych ilości jonów, które mogą niekorzystnie wpływać na jakość uzyskiwanych wyników. Dotyczy to szczególnie analiz na niskich poziomach stężeń. W przypadku oznaczeń jonów na poziomach kilkunastu – kilkudziesięciu mg/dm3 wpływ ten będzie odpowiednio mniejszy.
O ile to możliwe zaleca się w danym okresie użytkowania chromatografu jonowego analizować próbki o zbliżonym składzie jonowym. Takie postępowanie uchroni nas przed koniecznością zakupu nowej kolumny, której koszt to wydatek kilku tysięcy złotych oraz popełnienia błędów w analizie ilościowej.
Podczas analizy próbek na poziomie śladów i ultraśladów konieczne jest zachowanie w laboratorium rygorystycznych zasad czystości i bezpieczeństwa. Dotyczy to m.in. stosowania odpowiednich rękawic i okularów. Pomieszczenie, w którym wykonywane są tego rodzaju analizy musi być wolne od nieorganicznych zanieczyszczeń i nie wolno stosować w nim np. stężonych kwasów czy zasad.
Również pojemniki z próbką umieszczone w automatycznym podajniku próbek powinny być zabezpieczone odpowiednim zamknięciem (np. kapsel z tworzywa sztucznego). Naczynia na próbki nigdy nie powinny być czyszczone za pomocą kwasów, zasad lub mydeł.
Najtrudniejszy pierwszy krok, czyli zaczynamy pracę z chromatografem jonowym
Po włączeniu chromatografu jonowego i uruchomieniu pompy należy wykondycjonować układ, co związane jest z jego przemywaniem eluentem przez około 10-30 minut, aż do uzyskania stabilnej linii bazowej. Prawidłowe przygotowanie chromatografu jonowego do pracy gwarantuje uzyskiwanie powtarzalnych wyników analizy. Przykładowe chromatogramy 10 próbek wzorcowych anionów przedstawiono na rysunku 1.
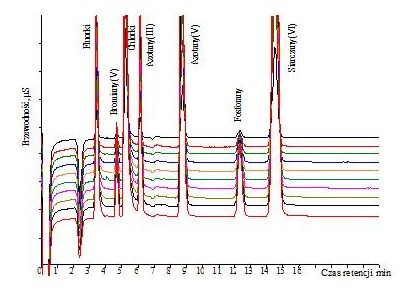
Rys.1. Chromatogramy roztworów wzorcowych anionów
Kolumna – Metrohm Metrosep A Supp 5
Eluent – 3,2 mM Na2CO3 + 1,0 mM NaHCO3
Natężenie przepływu eluentu – 0,7 ml/min
Detekcja – konduktometryczna z tłumieniem przewodnictwa
Świeżo przygotowane eluenty można wykorzystywać przez kilka kolejnych dni. Z prostych obliczeń wynika, że przy natężeniu przepływu eluentu 1,0 ml/min x 8 godzin daje 480 ml, czyli butelka z eluentem o pojemności 2 000 ml powinna wystarczyć na około 4 dni robocze.
Jeżeli chromatograf jonowy pracuje dłużej w ciągu doby (np. jest wyposażony jest w automatyczny podajnik próbek) to zużycie eluentu będzie odpowiednio szybsze.
Rozwój bakterii i glonów w układzie może spowodować poważne szkody w postaci niestabilnej linii podstawowej i konieczności długiego i żmudnego oczyszczania całego układu. To niekorzystne zjawisko zależy m.in. od rodzaju stosowanych eluentów i próbek, oraz od temperatury i nasłonecznienia pomieszczenia, w których pracuje chromatograf. W celu zapobiegania rozwojowi glonów lub bakterii do eluentu można dodać niewielką ilość (np. 2%) metanolu lub acetonu. Dodatek rozpuszczalników organicznych poprawia ponadto kształt pików i przyśpiesza ich wymywanie. Pamiętać jednak należy, że rozpuszczalniki organiczne mogą przeszkadzać w prawidłowej pracy niektórych supresorów i kolumn analitycznych.
Objętość zastosowanej pętli wstrzykowej powinna stanowić kompromis pomiędzy granicami wykrywalności jonów analitu, a wpływem matrycy próbki na jakość rozdzielania oraz żywotność kolumny analitycznej. Dla rutynowych oznaczań (np. wody do spożycia, wody opadowe) zaleca się, aby objętość ta była raczej niewielka (np. 20 μl). W określonych przypadkach (np. oznaczanie śladów BrO3–) zalecane jest stosowanie pętli o większej objętości np. 1000 µL. Kompromis polega na tym, że duża objętość nastrzyku powoduje wprowadzenie do kolumny nie tylko większej ilości jonów analitu (co jest korzystne ponieważ poprawia wykrywalność), ale także większej ilości zanieczyszczeń, co definitywnie nie sprzyja utrzymaniu właściwej charakterystyki kolumny.
Jedną z zalet chromatografii jonowej jest możliwość rozdzielania i oznaczania zarówno anionów, jak i kationów. W praktyce ten sam chromatograf po zmianie kolumn i eluentu stosowany jest zamiennie do analizy anionów lub kationów. Postępowanie takie jest oczywiście dopuszczalne, niemniej należy pamiętać o konieczności przemycia całego układu odpowiednim eluentem zanim podłączy się właściwą kolumnę do danego rodzaju analiz.
W chromatografii jonowej, tak jak i w innych metodach analitycznych istotna jest wstępna wiedza o rodzaju próbki będącej przedmiotem analizy, spodziewanym zakresie stężeń oraz rodzaju matrycy. Informacje takie często są dostępne, a gdy ich uzyskanie nie jest możliwe (np. związane jest to z utajnieniem próbki w procesie akredytacji) wtedy zalecana jest wstępna analiza za pomocą prostszych metod, np. testów paskowych. Wstępne cenne informacje o składzie jonowym próbki można uzyskać przeprowadzając pomiar jej przewodnictwa właściwego oraz pH.
Optymalizacja rozdzielania, czyli co zrobić kiedy dysponujemy tylko jedną kolumną analityczną
Optymalizacja procesu rozdzielania to dobór najbardziej odpowiedniej dla danej próbki warunków analitycznych, czyli: kolumny analitycznej, rodzaju i stężenia eluentu, jego pH i natężenia przepływu, a także rodzaju detektora i parametrów jego pracy. Niestety często nawet te możliwości są ograniczone, np. gdy laboratorium dysponuje tylko jedną kolumną analityczną i wyłącznie detektorem konduktometrycznym. W takim przypadku pole manewru ograniczone jest do zmian stężenia eluentu i/lub natężenia jego przepływu.
Zakres stężeń poszczególnych jonów w analizowanych próbkach nie powinien przekraczać kilkudziesięciu – kilkuset (w przypadku głównych jonów) mg/dm3 i zależy od rodzaju stosowanej kolumny analitycznej. Optymalne dla danej kolumny warunki analityczne takie jak: rodzaj, stężenie i natężenie przepływu eluentu wraz z przykładowym chromatogramem są zazwyczaj podane w materiałach dostarczonych przez producenta kolumny. W określonych przypadkach (np. gdy stężenie jednego z jonów znacznie przekracza stężenia eluowanych w zbliżonym czasie retencji innych jonów) należy przeprowadzić optymalizację procesu rozdzielania. Dotyczy to przede wszystkim par takich jonów jak: BrO3–/Cl–; Cl–/NO2– czy Na+/NH4+. Związane jest to ze zmianą stężenia eulentu, jego pH, natężeniem przepływu, a czasami konieczna jest zmiana kolumny rozdzielającej na dedykowaną do danego rodzaju oznaczeń, lub zmiana rodzaju eluentu.
Dobór właściwych warunków rozdzielania (rodzaju wypełnienia kolumny; rodzaju, stężenia i natężenia przepływu eluentu, rodzaju supresji i detekcji) uzależniony jest od wielu czynników (m.in. od rodzaju oznaczanych jonów, ich trwałości i stężeń oraz wartości pH próbki). Niektórzy producenci aparatury do chromatografii jonowej oferują pomoc w optymalizacji procesów rozdzielania, w postaci programów komputerowych (np. „Virtual Column” firmy Dionex).
Programy takie są pomocne w optymalnym doborze składu fazy ruchomej i warunków rozdzielania, a także doboru pH eluentu, stężenia modyfikatorów oraz siły jonowej eluentu. Możliwy jest też komputerowy dobór rodzaju i wymiarów kolumny, wielkości cząstek wypełnienia i przepływu fazy ruchomej w celu uzyskania optymalnego dla danych warunków rozdzielania analitów.
Ponad 20-letnie doświadczenia i kontakty z pracownikami laboratoriów rutynowo wykonujących analizy wód i ścieków z wykorzystaniem chromatografii jonowej przekonują mnie, że w laboratoriach tych najczęściej wykorzystuje się standardowe warunki analityczne i rekomendowane przez producentów kolumny.
Najpopularniejszym eluentami stosowanym w chromatografii jonowej z tłumieniem przewodnictwa do rozdzielania i oznaczania nieorganicznych anionów są wodne roztwory węglanów i wodorowęglanów sodu. Są to eluenty tanie i bezpieczne w użytkowaniu. Niemniej nawet niewielki błąd w odważeniu odpowiedniej odważki Na2CO3 lub NaHCO3 może skutkować istotnymi zmianami w czasach retencji poszczególnych jonów. Przykłady wpływu stężenia eluentu na czasy retencji jonów przedstawiono na rysunku 2.
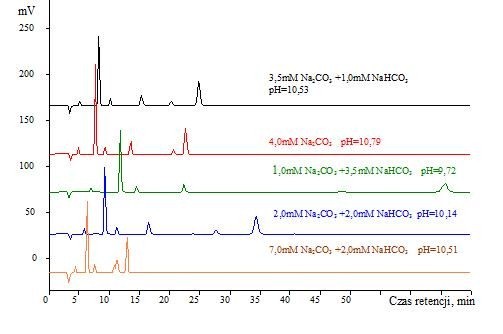
Rys. 2. Chromatogramy rozdzielania próbki wzorcowej nr 1 z wykorzystaniem kolumny Metrohm A Supp 5 i eluentów Na2CO3/NaHCO3 o różnych stężeniach
Z powyższego rysunku wynika, że w zależności od zawartości węglanu i/lub wodorowęglanu w eluencie oraz ich wzajemnym stosunku – czasy retencji rozdzielanych nieorganicznych anionów są silnie zróżnicowane. I tak dla eluentu 3,5 mM Na2CO3 +1,0 mM NaHCO3 o pH=10,53 całkowity czas wymywania wynosi około 25 minut, a piki poszczególnych anionów są dobrze rozdzielone i wykształcone. Zmiana eluentu na 4,0 mM Na2CO3 skraca całkowity czas rozdzielania, ale różnice w czasach retencji jonów PO43- i SO42- są na tyle niewielkie, że w przypadku próbki o dużej zawartości jednego (lub obydwu) tych jonów mogą one nie być dobrze rozdzielone.
Interesujące zmiany dotyczą eluentu o składzie 1,0 mM Na2CO3 +3,5 mM NaHCO3 (pH=9,72). Jony SO42- są wymywane dopiero po ponad 70 minutach i są oczywiście bardzo dobrze rozdzielone wobec poprzedzających je jonów PO43-. Pytanie tylko czy tak długie czasy retencji są uzasadnione ekonomicznie, szczególnie podczas rutynowych analiz? Sytuacja ta i analogiczna dla eluentu o składzie 2,0 mM Na2CO3 +2,0 mM NaHCO3 stanowi potwierdzenie, że nawet relatywnie niewielkie zmiany eluentu mogą istotnie zmieniać czasy retencji. Może to być bardzo korzystne dla specyficznych rodzajów oznaczeń i matryc próbek. Przykładowo, jeżeli analizujemy próbki o bardzo dużej zawartości np. jonów SO42- i śladowej zawartości jonów np. NO3– – dysponując np. kolumną Metrohm A Supp 5 i eluentem o składzie 2,0 mM Na2CO3 +2,0 mM NaHCO3 możliwe jest jednoczesne rozdzielanie i oznaczanie tych i innych jonów bez specjalnego przygotowania próbki np. w skomplikowanych procesach usuwania nadmiaru jonów dominujących; zatężenia jonów występujących na niskich poziomach stężeń lub rozcieńczania próbki.
Ostatni chromatogram przedstawiony na rysunku 2 to chromatogram uzyskany dla eluentu o składzie 7,0 mM Na2CO3 +2,0 mM NaHCO3, który to eluent okazał się być zbyt mocny, aby właściwie jednocześnie rozdzielać i oznaczać wszystkie główne nieorganiczne aniony. Porównując ten chromatogram z chromatogramem uzyskanym dla eluentu o dwukrotnie mniejszym stężeniu wyraźnie widać jak bardzo skład eluentu wpływa na czasy retencji rozdzielanych jonów, nawet jeżeli ich pH jest takie samo.
Zaleca się odgazowywanie eluentów, co zapobiega deformacji linii podstawowej i niestabilnej pracy systemu. Proces ten przeprowadzana się zazwyczaj poprzez przepuszczenie strumienia gazu obojętnego (np. helu) przez roztwór eluentu przez kilkanaście minut. Można to wykonać również za pomocą pompki wodnej, pompy próżniowej albo łaźni ultradźwiękowej. Nowoczesne chromatografy jonowe posiadają degazery wbudowane w przyrządzie.
Na rysunku 3 przedstawiono chromatogramy rozdzielania anionów z i bez użycia absorbera CO2.
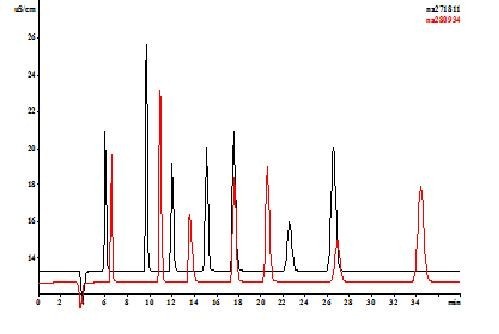
Rys.3. Wpływ absorbera CO2 na czasy retencji głównych nieorganicznych anionów.
Warunki analitczne:
Kolumna – Metrohm Metrosep Supp 5
Eluent – 3,2 mM Na2CO3 + 1,0 mM NaHCO3
Objętość nastrzyku – 25 µl
Detekcja – konduktometryczna z tłumieniem przewodnictwa
Absorber – Metrohm MCS
Brak absorbera CO2 powoduje w powyższym wypadku wzrost czasów retencji wszystkich jonów, a dla jonów SO42- różnica ta wynosi ponad 8 minut.
Wpływ temperatury na jakość oznaczania w chromatografii jonowej nie jest tak istotny jak w innych metodach chromatograficznych, aczkolwiek może być znaczący. Nowoczesne chromatografy jonowe wyposażone są w termostaty nie tylko detektora konduktometrycznego, ale również kolumn.
Wpływ temperatury na jakość rozdzielania jest uzależniony od rodzaju wypełnienia kolumny. Ogólnie można stwierdzić, że wzrost temperatury powoduje przyśpieszenie wymywania jonów jednowartościowych i wydłużenie czasów retencji jonów wielowartościowych, tak jak przedstawiono to na rysunku 4.
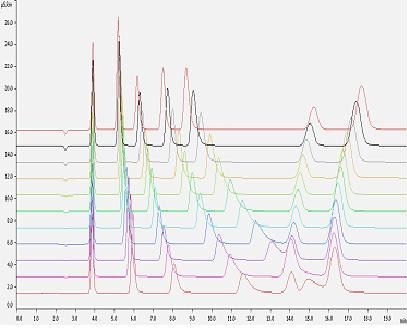
Rys.4 . Wpływ temperatury na szybkość wymywania głównych nieorganicznych anionów
Warunki rozdzielania:
Kolumna anionowymienna – Metrohm A Metrosep Sup 5
Eluent – 3,5 mM Na2CO3 + 3,0 mM NaHCO3,
Natężenie przepływu eluentu – 0,7 ml/min,
Detekcja – konduktometryczna z tłumieniem przewodnictwa
W niektórych wypadkach np. kolumny Metrohm Metrosep 7 wraz ze zmianą temperatury może dojść do zmiany kolejności wymywania anionów, szczególnie jonów NO3– i PO43-.
Analiza jakościowa i ilościowa, czyli co i ile mamy w próbce
Po optymalizacji procesu rozdzielania kolejnym ważnym etapem analizy jest sporządzenie roztworów wzorcowych do kalibracji przyrządu. Obecnie dostępnych jest na rynku wiele gotowych mieszanin wzorców odpowiednich jonów. Zakupując takie mieszaniny musimy pamiętać, że nie mamy możliwości wpływu na wzajemne stosunki stężeń oznaczanych jonów, w związku, z czym w wielu przypadkach wygodniej i bezpieczniej jest stosować wzorce pojedynczych jonów.
Stężenia poszczególnych jonów w roztworach do kalibracji powinno mieścić się w połowie spodziewanego zakresu ich stężeń w analizowanych próbkach. Ponadto zakres ich stężeń nie powinien przekraczać 3 rzędów wielkości, czyli lepiej przygotować dwie krzywe kalibracyjne np. w zakresie 0,5 – 5,0 mg/dm3 oraz 5,0 – 50,0 mg/dm3, niż jedną serię w zakresie od 0,5 do 50,0 mg/dm3, ponieważ zależność uzyskiwanych sygnałów od stężenia jonów analitu może nie być liniowa w tak szerokim zakresie stężeń.
Analiza jakościowa i ilościowa z zastosowaniem chromatografii jonowej oparta jest na tych samych zasadach, jakie obowiązują w innych metodach chromatograficznych. Analiza jakościowa odbywa się na podstawie porównania czasów retencji jonów analitu w próbce wzorcowej i próbce badanej uzyskanych w identycznych warunkach chromatografowania. W jednakowych warunkach analitycznych chromatografowana substancja może mieć nieznacznie zróżnicowane czasy retencji, co może być związane m.in. ze zużyciem kolumny oraz pH i rodzajem matrycy analizowanej próbki.
Analiza ilościowa przeprowadzana jest na podstawie pomiaru wysokości lub pola powierzchni pików. Pamiętać należy, że wielkości pików różnych substancji o tych samych stężeniach są zwykle różne, co spowodowane jest ich różną wykrywalnością przez dany detektor.
Podstawowe sposoby chromatograficznej analizy ilościowej są następujące: sposób kalibracji bezwzględnej (tzw. metoda wzorca zewnętrznego), sposób normalizacji wewnętrznej, sposób z wzorcem wewnętrznym oraz jego odmiana, w której wzorcem jest substancja oznaczana (sposób z dodatkiem substancji oznaczanej).
Sposób przeprowadzenia kalibracji zależy od następujących czynników: rodzaju przyrządu pomiarowego, ilości próbek, wymaganej dokładności oznaczenia, składu matrycy próbki oraz możliwości zmiany składu próbki w trakcie procesu analitycznego.
Piki, które nie cieszą
W chromatografii jonowej z tłumieniem przewodnictwa, na początku chromatogramu tuż przed pikami jonów fluorkowych pojawia się tzw. pik nastrzykowy. Jest to pik o polaryzacji ujemnej, a jego obecność związana jest z faktem, że podczas przepływu eluentu przez kolumnę analityczną, jony eluentu oddziaływają z grupami funkcyjnymi w wymieniaczu jonowym. W momencie wstrzyknięcia próbki proces ten zostaje przerwany, co powoduje pojawienie się piku nastrzykowego na chromatogramie. Pik ten może wpływać na jakość oznaczania jonów najszybciej wymywanych z kolumny (fluorki, mrówczany, octany), a w skrajnych przypadkach, wręcz uniemożliwiać ich oznaczanie.
Uwagi te dotyczą przede wszystkim kolumn rozdzielających starszej generacji. Nowoczesne wysokoselektywne kolumny anionowymienne charakteryzują się dobrym rozdzielaniem jonów F– wobec piku nastrzykowego i jonów Cl–. Dla tych kolumn różnice w czasach retencji jonów F– i Cl– są na tyle duże, że kolumny te umożliwiają jednoczesne rozdzielanie jonów F–, ClO2–, BrO3– i Cl–, które euluują z kolumny rozdzielającej w tej właśnie kolejności. Jeżeli na chromatogramie pojawią się inne dodatkowe niezidentyfikowane piki pomiędzy pikami odpowiadającymi jonom F– i Cl–, a przedmiotem analiz nie są próbki wody chlorowanej (ClO2–) lub ozonowanej (BrO3–) to pochodzą one najprawdopodobniej od jonów niskocząsteczkowych kwasów karboksylowych (np. HCOO–, CH3COO–).
Ich jednoznaczna identyfikacja możliwa jest z zastosowaniem n.p. detektora spektrometrii mas, ale często w warunkach rzeczywistych musi wystarczyć użycie odpowiednich wzorców oraz „intuicja” i doświadczenie analityka związane z wiedzą o pochodzeniu badanej próbki.
W chromatografii jonowej bez tłumienia przewodnictwa na chromatogramie mogą pojawić się dwa piki nie pochodzące od jonów próbki. Pierwszy z nich jest pikiem nastrzykowym. Rozmiary drugiego piku, tzw. piku systemowego zależne są od pH, rodzaju i stężenia eluentu, stężenia jonów próbki, jej objętości oraz pH, jak również zastosowanego detektora.
Jeżeli pH próbki jest niższe od pH eluentu, jony eluentu (np. benzoesanu powszechnie stosowanego w chromatografii jonowej z tłumieniem przewodnictwa) są protonowane, a stężenie niezdysocjowanego benzoesanu w eluencie rośnie i jest on absorbowany na fazie stacjonarnej.
Ta część benzoesanu, która nie jest zaabsorbowana przechodzi przez kolumnę i jest rejestrowana w postaci piku systemowego. Wprowadzenie do układu próbki o pH wyższej niż pH eluentu zwiększa stopień dysocjacji benzoesanu i obniża wielkość piku systemowego. Piki takie występują również w chromatografii jonowej z tłumieniem przewodnictwa, jednak ich wpływ na jakość analiz jest niewielki, ponieważ rozmiary pików jonów oznaczanych (przy tych samych stężeniach) są zdecydowanie większe niż w chromatografii jonowej bez tłumienia przewodnictwa.
Pik systemowy jest zawsze obecny, gdy stosuje się eluenty węglanowe, a czas jego retencji zależny jest od rodzaju wypełnienia w kolumnie analitycznej. Położenie piku systemowego (węglanowego) na chromatogramie dla wybranych kolumn anionowymiennych firmy Metrohm przedstawiono na rysunku 5.
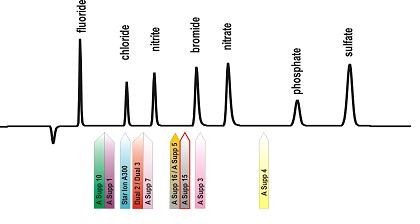
Rys. 5. Położenie piku systemowego dla wybranych kolumn anionowymiennych firmy Metrohm
W chromatografii wykluczania jonowego występują dwa piki systemowe – ujemny i dodatni. Pik ujemny obserwowany jest, gdy wprowadzona do kolumny analitycznej próbka zawiera mniej aktywnych jonów niż ich stężenie w eluencie.
Z kolei pik dodatni obserwowany jest, gdy wprowadzona do kolumny analitycznej próbka, zawiera więcej aktywnych jonów niż ich stężenie w eluencie. Gdy stężenie aktywnych składników eluentu jest takie samo jak w analizowanej próbce, pik systemowy nie pojawia się na chromatogramie.
Przechowywanie i regeneracja kolumn
Charakterystyka kolumny pogarsza się w wyniku zużycia. W efekcie tego skróceniu ulegają czasy retencji poszczególnych jonów, a kształty ich pików są coraz bardziej rozmyte i niesymetryczne. Konieczna staje się wtedy wymiana przedkolumny i kolumny analitycznej na nową, lub jej regeneracja, polegająca na przemyciu w przeciwprądzie odpowiednimi roztworami, zgodnie z zaleceniami producenta. W zależności od rodzaju analizowanych próbek kolumna analityczna może wymagać wymiany lub regeneracji po około 100 do nawet 1000 analiz. Decyzja o tym, kiedy należy wymienić kolumnę analityczną należy do analityka i zazwyczaj uzależniona od rodzaju analizowanych próbek, wymaganej precyzji oznaczeń oraz …….. możliwości finansowych jednostki.
Nie można dopuścić do wyschnięcia złoża kolumny podczas jej dłuższego przechowywania. W takich wypadkach kolumnę przemywa się stężonym eluentem i zatyka koreczkami.
Ogólnie, aby właściwie korzystać z kolumn analitycznych należy:
- Chronić ją za pomocą przedkolumn, które należy systematycznie wymieniać.
- Używać kolumny zgodnie z zaleceniami producenta (pH, natężenie przepływu, ciśnienie, rozpuszczalniki organiczne).
- Stosować ultra czystą wodę do przygotowania eluentów.
- Unikać rozwoju bakterii i wtrącania się osadów.
- Unikać wysuszenia i szoku termicznego/mechanicznego.
- Unikać przeładowania kolumny.
- Filtrować każdą próbkę.
- Stosować kolumienki do przygotowania próbek tam, gdzie to konieczne.
Najważniejsze problemy analityczne związane z użytkowaniem chromatografów jonowych i metody ich rozwiązywania
Główne problemy związane z wykonywaniem analiz za pomocą chromatografu jonowego dotyczą zazwyczaj następujących zagadnień:
- Zmian czasów retencji jonów.
- Niestabilności linii bazowej.
- Wielkości rejestrowanego sygnału.
- Zmian ciśnienia.
- Zmian przewodnictwa tła.
- Kształtów uzyskiwanych pików chromatograficznych.
Problemy związane z czasami retencji analitów dotyczą ich skracania się, wydłużania lub nieregularnych zmian. Skracanie się czasów retencji związane jest najczęściej z niewłaściwym (zbyt mocnym) eluentem lub zwiększonym natężeniem jego przepływu. Skracanie się czasów retencji może być również związane z absorbcją CO2 w eluencie i zmianą jego pH. Z kolei wydłużanie czasów retencji najczęściej dotyczy zbyt niskiego stężenia eluentu w stosunku do deklarowanego i/lub niższego natężenia przepływu. Z kolei zmiany/fluktuacje czasów retencji dotyczą najczęściej wycieków w układzie, zapowietrzeniu pompy eluentu lub absorbcją CO2. Konieczne jest wtedy odgazowanie eluentu i usunięcie bąbelków powietrza z pompy.
Również problemy związane z szumami linii bazowej dotyczą zazwyczaj zapowietrzonej pompy i/lub nieodgazowanego eluentu. Na wielkość szumów wpływa temperatura, w związku z czym chromatograf powinien pracować w stabilnych warunkach środowiskowych, a temperatura detektora powinna być o 5ºC wyższa od temperatury otoczenia.
Inne przyczyny wysokich szumów to m.in. zanieczyszczona kolumna analityczna lub filtr próbki, które należy oczyścić lub wymienić. W wypadku stosowania detektora amperometrycznego należy pamiętać o okresowym czyszczeniu elektrody roboczej.
Problemy związane z rejestrowanym pikiem objawiają się najczęściej w postaci braku sygnału, jego zbyt dużej wielkości, ogonowaniu lub obcinaniu pików. Należy sprawdzić połączenia kapilar do zaworu wstrzykowego. Inne przyczyny takiej sytuacji mogą być równie trywialne, np. brak określonych połączeń, wycieki w układzie czy niewłaściwie dobrany eluent lub zakres pracy detektora.
Jeżeli rejestrowany sygnał jest zbyt niski lub bardzo wysoki związane to może być m.in. ze zbyt małą objętością wprowadzonej próbki, niewłaściwą pozycją zaworu wstrzykowego lub niewłaściwymi parametrami pracy detektora.
Ucinanie piku (zbyt wysoki sygnał) powodowany jest za dużym stężeniem analitów w stosunku do pojemności wymiennej kolumny. W takim wypadku należy zastosować mniejszą objętość próbki (mniejszą pętlę wstrzykową), rozcieńczyć próbkę do właściwego zakresu stężeń, sprawdzić ustawienia zakresu pomiarowego i pełnej skali pracy detektora podczas rejestracji chromatogramu.
Ponadto w wypadku rozdzielania kationów nakładanie się pików może być spowodowane niewłaściwym pH próbki. Należy pamiętać, ze wzorce kationów do metod spektroskopowych (np. ASA, ICP) czy metod woltamperometrycznych nie są właściwe do stosowania w chromatografii jonowej.
Problemy związane z ciśnieniem w układzie chromatograficznym dotyczą zazwyczaj; jego zbyt wysokiej wartości, braku ciśnienia lub bardzo niskiej wartości oraz jego nieregularnych zmian (skoki i spadki wartości). Jeżeli ciśnienie jest zbyt wysokie należy zlokalizować miejsce zapchania toru analitycznego i sprawdzić przepływ eluentu w pełnym zakresie. Inną przyczyny wzrostu ciśnienia może być np. zastosowanie niewłaściwego dodatku rozpuszczalnika organicznego i kolumny analitycznej niewłaściwej dla danego rozpuszczalnika.
Z kolei brak ciśnienia lub jego fluktuacja w układzie związane jest najczęściej z wyciekami, zapowietrzeniem pompy i/lub eluentu. Większość czynności związanych z ustaleniem tej przyczyny oraz ich usunięcia może być wykonana przez osobę obsługującą przyrząd. W wypadku, gdy konieczne jest czyszczenie głowicy pompy, sprawdzenie i wymiana zaworów, uszczelnianie tłoków pompy itp. konieczne jest wezwanie serwisu.
Podsumowanie i wnioski
Codziennie w tysiącach laboratoriów kontrolno-pomiarowych, jak i naukowo-badawczych na całym świecie wykonuje się miliony oznaczeń substancji jonowych w różnego rodzajach próbek ciekłych, stałych i gazowych. Zakres jakościowy i ilościowy tych analiz zazwyczaj jest bezpośrednio związany z wiedzą toksykologiczną i co z niej wynika – z wymaganiami światowymi, krajowymi lub lokalnymi.
Do najczęściej rutynowo analizowanych próbek należą próbki wód i ścieków w zakresie oznaczań metali oraz nieorganicznych anionów i kationów. Metodą, która ze względu na swoje zalety takie, jak szybkość analiz, ich dokładność powtarzalność oraz możliwość pełnej automatyzacji systematycznie zastępuje dotychczas stosowane metody klasyczne jest chromatografia jonowa.
Od oficjalnej daty powstania chromatografii jonowej, tj. roku 1975 – metoda ta ewoluowała od prostej metody oznaczania głównych nieorganicznych anionów i kationów w wodach, do wyrafinowanej metody separacji umożliwiającej w połączeniu z nowoczesnymi detektorami oznaczać śladowe ilości substancji w różnego rodzajach próbek ciekłych, stałych i gazowych [15].
W ostatnich latach ogromne zmiany wprowadzono w zakresie wypełnień w kolumnach jonowymiennych, co spowodowało rozszerzenie rodzajów stosowanych eluentów i metod detekcji. Wielkie zmiany dotyczą również metod przygotowania próbek do analizy oraz nowych rodzajów matryc i analitów oznaczanych tą metodą [16, 17].
W zależności od wyposażenia koszt chromatografu jonowego wynosi od około 60 000 zł do 400 000 zł, aczkolwiek fantastyczne narzędzie badawcze jakim jest zestaw chromatograf jonowy z detektorem ICP-MS lub MS to już cena zdecydowanie wyższa. Na tym poziomie (i wyższym) kształtują się również ceny chromatografów gazowych czy spektrometrów absorpcji atomowej – urządzeń powszechnie stosowanych w laboratoriach badawczych i usługowych. Czy zatem „koń roboczy” jakim jest chromatograf jonowy z detektorem konduktometrycznym to dobra inwestycja? Na pewno tak. Należy ponadto pamiętać, że w pełni zautomatyzowany chromatograf jonowy pozwala oznaczać kilkadziesiąt próbek dziennie w zakresie kilku – kilkunastu jonów, co ma szczególne znaczenie podczas analiz rutynowych. Poza tym bieżące koszty użytkowania chromatografu jonowego są zdecydowanie niższe niż np. chromatografów HPLC lub GC.
Chromatograf jonowy użytkowany w prawidłowy sposób przez doświadczonego i świadomego jego zalet i ograniczeń analityka, stanowić powinien bardzo ważny, wiarygodny i wydajny element systemu analitycznego w laboratoriach kontrolno-pomiarowych, jak i naukowo-badawczych.
Biorąc pod uwagę postępy w zakresie chromatografii jonowej oraz zakres ich zastosowań przyrządy te w pewnym sensie upodabniają się do jonometrów. Wynika to z faktu, że nowoczesne chromatografy jonowe zaopatrzone są w automatyczne generatory eluentu, automatyczne podajniki próbek oraz programy umożliwiające automatyczne rozcieńczanie próbek i kalibrację przyrządów [18].
Chromatografia jonowa, która została opracowana w USA w latach 70-tych XX wieku, obecnie zdobywa sobie nowe rynki nie tylko w krajach wysokouprzemysłowionych, ale i w krajach, które dopiero wchodzą na ścieżkę szybkiego rozwoju, takie jak kraje Europy Centralnej i Środkowej [19].
Nie sposób w tak krótkim artykule opisać wszystkie problemy związane z użytkowaniem chromatografu jonowego i metody ich rozwiązywania. Najlepszym wyjściem jest nauka na własnych błędach („trening czyni Mistrza”), lub udział w praktycznych warsztatach z chromatografii jonowej, np. takich jak organizowane od kilku lat przez firmę Metrohm Polska (www.metrohm.pl).
Literatura
[1] Tswett M.S., Physikalisch-Chemische Studien Uber das Chlorophyll. Die Adsorptionen, Ber.Bot.Ges., 24, (1906), 316–332.
[2] Lucy C.A., Evolution ofIion-exchange: from Moses to the Manhattan Project to Modern Times, J.Chromatogr., 1000, (2003), 711-724.
[3] Small H., Stevens T.S., Bauman W.C., Novel Ion Exchange Chromatographic Method Using Conductometric Detection, Anal. Chem., 47, (1975), 1801–1886.
[4] Michalski R., Chromatografia jonowa. Podstawy i zastosowania. WN-T, Warszawa, 2005.
[5] Michalski R., Chromatografia jonowa – zalety i ograniczenia, Laboratorium, 3, (2005), 27–31.
[6] Łyko A., Michalski R., Oznaczanie jonów w chłodziwach, kąpielach myjących oraz emulsjach, LAB, 15, (2010), 36-38
[7] Michalski R., Nieorganiczne utlenione halogenopochodne uboczne produkty dezynfekcji w wodach do picia – powstawanie, oznaczanie, regulacje prawne, Ekologia i Technika, 2/68, (2004), 40–49.
[8] Michalski R., Bromiany(V), chlorany(III) i chlorany(V) w wodach do picia, LAB, 3, (2006), 6–11.
[9] Michalski R., Wykorzystanie chromatografii jonowej w analityce specjacyjnej nieorganicznych jonów, LAB, 4, (2006), 6–10.
[10] Michalski R., Zastosowania chromatografii jonowej w badaniach próbek medycznych, przemysłowych i spożywczych, Laboratorium, 5, (2005), 28–32.
[11] Michalski R., Chromatografia jonowa, jako referencyjna metoda oznaczania nieorganicznych jonów w wodach i ściekach, LAB, 2, (2006), 30–35.
[12] Michalski R., Chromatografia jonowa – uwagi użytkownika, Laboratorium, 3, (2005), 31-35.
[13] Michalski R., Metody przygotowania próbek w chromatografii jonowej, Laboratorium, 4, (2005), 23–25.
[14] Michalski R., Bromiany (metoda chromatografii jonowej) [w] Uboczne produkty dezynfekcji wody, pod redakcją J.Dojlido, Seria: Wodociągi i Kanalizacja, Monografie, Polskie Zrzeszenie Inżynierów i Techników Sanitarnych, Wyższa Szkoła Ekologii i Zarządzania w Warszawie, Warszawa 2002, str.139-158.
[15] Michalski R., Oznaczanie gazowych zanieczyszczeń powietrza metodą chromatografii jonowej, LAB, 2, (2007), 16-18.
[16] Michalski R., Chlorany(VII) – nowe, niedocenione zagrożenie środowiska, LAB, 3, (2007), 16–19.
[17] Michalski R., Nowoczesne metody i techniki analityczne. Problemy i wyzwania, Laboratorium, 3, (2009), 10-14
[18] Michalski R., Podstawy chromatografii jonowej, SWSZ, Katowice, 2011.
[19] Michalski R., Environmental Applications of Ion Chromatography in Eastern and Central Europe, J. Chromatogr. Sci., 48/7, (2010), 559-565.
*Rajmund Michalski
Instytut Podstaw Inżynierii Środowiska PAN