Analityka pozostałości farmaceutyków w żywności i próbkach środowiskowych
Autor: Anna Białk, Piotr Stepnowski*
Dzięki ogromnym nakładom przemysłu farmaceutycznego przy jednoczesnym postępie nauki i rozwoju technik badawczych osiągnięto bardzo wiele korzyści w dziedzinie farmaceutycznej ochrony zdrowia i życia ludzkiego. Jednakże mówiąc o osiągnięciach w dziedzinie farmakoterapii, należy także pamiętać o wielu problemach jakie do tej pory nie zostały rozwiązane w tym, przede wszystkim, o przedostawaniu się pozostałości środków leczniczych do środowiska i żywności [1].
Farmaceutyki są substancjami aktywnymi biologicznie, które znajdują swe zastosowanie w medycynie człowieka, a także weterynarii. Stosowane są przede wszystkim w celach leczniczych i profilaktycznych, lecz nie tylko. Znajdują bowiem powszechne zastosowanie w hodowli zwierząt – produkcji żywności oraz jako tak zwane stymulatory wzrostu. W przeszłości, w celach hodowlanych regularnie aplikowano niedozwolone obecnie antybiotykowe stymulatory wzrostu oraz inne leki weterynaryjne. Priorytetem było wówczas zapewnienie odpowiednich przyrostów masy żywego inwentarza, nie dbając o kwestie związane z jakością żywności i bezpieczeństwem uzyskiwanych surowców i produktów żywnościowych pochodzenia zwierzęcego. Problem ten zyskał już wymiar globalny i obecnie zgodnie z regulacjami Komisji Europejskiej (Dyrektywa Rady 96/23/EC oraz Decyzja Komisji 93/256/EEC), do krajów członkowskich należy kontrola jakości żywności. Ponadto, wszystkie Państwa Członkowskie zostały zobowiązane do wycofania z dniem 1 stycznia 2006 roku dotychczas stosowanych w charakterze dodatku do pasz, antybiotykowych stymulatorów wzrostu i ograniczenia się do stosowania tylko substancji dozwolonych, wobec których mają także obowiązek prowadzenia kontroli [2-5].
Po zaaplikowaniu do ustroju, część farmaceutyku działa leczniczo i ulega metabolizmowi, pozostałość zaś (w zależności od rodzaju farmaceutyku od 10 do 90%) zostaje wydalona praktycznie w niezmienionej formie. Wydalane są także produkty I i II fazy metabolizmu, które po przedostaniu do środowiska mogą ulec ponownemu przekształceniu w formę aktywną [4,6-7]. Ponadto, zdarza się, iż metabolity I fazy są bardziej reaktywne i toksyczne niż związek macierzysty. Pomimo, iż w przeciągu kilku ostatnich lat bardzo wiele uwagi poświęcono źródłom emisji jak i losowi środowiskowemu pozostałości farmaceutyków wciąż niewiele jest informacji na temat ich rzeczywistego zagrożenia jakie stwarzają dla środowiska i zdrowia człowieka. Wciąż niewyjaśnione są dokładnie zależności pomiędzy obecnością w środowisku oraz w żywności antybiotyków i ich metabolitów a zjawiskiem lekooporności, choć z drugiej strony wiadomo, iż konsekwencje obecności lekoopornych szczepów bakterii stanowią i będą stanowić problem w wymiarze globalnym [4]. Aktualnie znaczna część leków stosowanych w produkcji mięsa gromadzi się w tkankach zwierzęcych, dlatego też istnieje podwyższone ryzyko przeniesienia tych substancji drogą układu pokarmowego do organizmu człowieka [2-3].
Wyróżnić można trzy zasadnicze drogi, którymi farmaceutyki i ich metabolity przedostają się do środowiska. Przede wszystkim, do emisji środków leczniczych dochodzi podczas ich produkcji przez przemysł farmaceutyczny. Ponadto środki te są zarzucane bez ich wcześniejszej utylizacji bezpośrednio do środowiska zarówno z gospodarstw domowych, jak i ze ściekami i odpadami szpitalnymi oraz wydalane są przez zwierzęta i ludzi. Leki wydalone przez człowieka trafiają do ścieków komunalnych, natomiast farmaceutyki weterynaryjne przedostają się do środowiska na skutek rozsiewania na polach obornika lub innych nawozów naturalnych, które zawierają odchody zwierząt. Można zatem uznać, iż wpływ leków weterynaryjnych na glebę i wody gruntowe jest znacznie większy niż na zanieczyszczenie wód powierzchniowych [4,6-11]. Przykładowo, szacuje się, iż ładunek antybiotyków dostających się do gleby wraz z nawozami sięga rzędu kilku kilogramów na hektar; stężenia oznaczanych antybiotyków przekraczają tam często 400 mg/kg gleby, przy czym największym udziałem charakteryzują się antybiotyki z grupy tetracyklin, stosowane często w hodowli trzody chlewnej [4]. Główne źródła pozostałości farmaceutyków w środowisku oraz drogi ich rozprzestrzeniania zostały przedstawione na rys. 1.
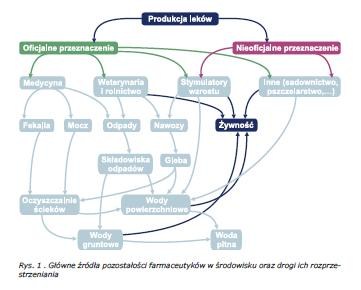
Pozostałości lekowe wraz ze ściekami komunalnymi trafiają do oczyszczalni ścieków, gdzie podlegają różnym procesom oczyszczania wód. W wyniku czego ulegać mogą całkowitej lub częściowej mineralizacji. Niestety, na podstawie wielu badań wynika, iż antybiotyki, środki hormonalne, niesteroidowe leki przeciwbólowe i przeciwzapalne oraz inne lecznicze substancje są odporne na procesy biodegradacji i nie podlegają całkowitej eliminacji [8-9, 12-15]. Substancje te i ich metabolity, które są trwałe i polarne nie będąc zatrzymywane ani też degradowane w oczyszczalni ścieków z łatwością migrują do środowiska wodnego i wraz z wodami oczyszczonymi odprowadzane są do wód powierzchniowych lub wraz z osadem ściekowym trafiają do gleby i wód gruntowych, na skutek stosowania tych osadów jako nawozów na polach uprawnych [7, 16]. Wody gruntowe i powierzchniowe są natomiast głównymi źródłami wody pitnej. Właściwości fizykochemiczne leków takie jak trwałość, odporność na biodegradację, duża rozpuszczalność w wodzie jak i niskie wartości współczynników sorpcji mogą także przyczyniać się do niskiego stopnia eliminacji tych związków podczas procesów uzdatniania wody przeznaczonej do celów konsumpcyjnych.
Farmaceutyki znajdujące się w środowisku mogą podlegać różnym procesom – ulegać biodegradacji, fotodegradacji lub też eliminacji na drodze hydrolizy czy sorpcji na cząstkach materii zawieszonej, gleby, osadach dennych i ściekowych [8-9].
Zdolność leków do adsorbowania się na innych cząstkach w dużej mierze zależy od ich zróżnicowanej budowy chemicznej, zawierającej ugrupowania o charakterze zasadowym jak i kwasowym. Z tego względu ich dystrybucja w środowisku wodnym bardzo silnie zależy od pH. Od odczynu środowiska zależeć będzie też zatem ich rozpuszczalność, hydrofobowość oraz współczynnik sorpcji [8, 10, 17].
Jednym z elementów sprzyjających usuwaniu tych związków ze środowiska jest także promieniowanie słoneczne. Spośród wielu farmaceutyków, przykładowo tetracykliny, chinoliny oraz sulfonamidy są substancjami wrażliwymi na światło. Zjawisko to ma istotne znaczenie w procesie samooczyszczania się wód powierzchniowych. Zależne jest od wielu czynników takich jak temperatura, intensywność światła jak i objętościowego natężenia przepływu wód [6-10].
Biodegradowalność farmaceutyków oraz ich toksyczność ostra wobec organizmów wodnych stanowią istotne czynniki wpływające na poziom oraz rozprzestrzenianie się tych substancji w środowisku. Biodegradacja tych związków może prowadzić do ich całkowitej mineralizacji lub do biotransformacji, czyli powstania nowych produktów mogących charakteryzować się dużo większą trwałością i wyższą toksycznością niż substancje macierzyste. Za procesy biodegradacji w ściekach komunalnych, wodach powierzchniowych i gruntowych odpowiedzialne są w największym stopniu bakterie, natomiast w przypadku zanieczyszczonych gleb znaczącą rolę w biodegradacji pełnią tam grzyby [18]. Przykładowy los środowiskowy farmaceutyków stosowanych przez człowieka został przedstawiony na rys. 2.
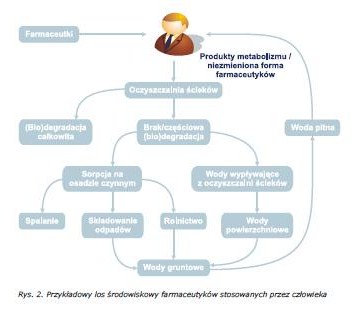
W związku z faktem, iż zastosowanie leków odgrywa coraz większą rolę w produkcji żywności, a także w związku z zagrożeniem jakie stanowią pozostałości farmaceutyków, ich metabolity oraz produkty degradacji w środowisku, oznaczanie farmaceutyków w poszczególnych elementach środowiska jak i żywności traktowane jest jako jedno z priorytetowych zadań z zakresu monitoringu środowiska oraz kontroli jakości żywności. Stało się konieczne podjęcie odpowiednich środków w celu ochrony konsumentów przed efektami pozostałości leków w żywności. Wciąż najważniejszym ograniczeniem jest dostępność odpowiednich procedur analitycznych, umożliwiających ilościowe oznaczenie zarówno pozostałości tych związków jak i ich metabolitów w próbkach środowiskowych i żywności, dzięki czemu możliwe będzie pełne oszacowanie rzeczywistego stopnia narażenia człowieka na te zanieczyszczenia [2-3, 17, 19]. Określenie zawartości śladowych ilości tak szerokiej gamy związków zróżnicowanych pod kątem chemicznym dodatkowo w próbkach o złożonym składzie matrycy jest poważnym wyzwaniem współczesnej analityki. Obecność farmaceutyków w środowisku stanowi także nowe wyzwanie dla technologii oczyszczania wód i ścieków, gdzie konieczny jest rozwój nowych technik skutecznie eliminujących omawiane związki [19].
Analityka zanieczyszczeń z grupy farmaceutyków jest bardzo trudnym zadaniem, ze względu na [20-21]:
- bardzo niskie stężenia tych związków rzędu kilku mg/kg lub ng/kg,
- różnorodność pod względem budowy chemicznej,
- konieczność oznaczania nie tylko związków pierwotnych, ale także produktów degradacji i metabolizmu,
- różnorodność matryc i różnice w poziomie ładunku zanieczyszczenia,
- możliwość występowania interferencji od innych składników próbki charakteryzujących się podobnymi właściwościami fizykochemicznymi,
- brak odpowiednich wzorców i materiałów odniesienia.
Oznaczenie pozostałości farmaceutyków w próbkach środowiskowych jak i żywności wymaga zastosowania bardzo czułych i selektywnych technik oznaczeń końcowych [2-3, 22]. W tym celu stosuje się głównie metody chromatograficzne, szczególnie chromatografię cieczową sprzęgniętą ze spektrometrią mas (LC-MS) lub tandemową spektrometrią mas (LC-MS/MS) [2-3, 5, 22-24].
Zakres zastosowania poszczególnych metod chromatograficznych w oznaczaniu omawianych związków został schematycznie przedstawiony na rys 3.
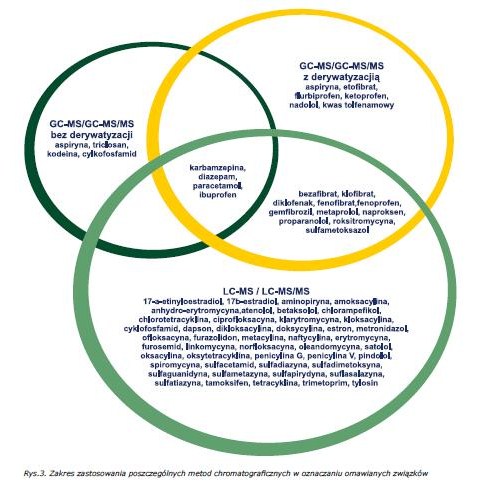
W procesie analitycznym pozostałości farmaceutyków każda czynność już od momentu pobierania próbki ma istotny wpływ na wynik końcowy oznaczenia [24-25]. Pobieranie próbek i ich przygotowanie do analizy zależą od matrycy w jakiej występują anality (ciekła lub stała). Próbki wodne pobierane są do szklanych nieprzezroczystych naczyń, w celu uniknięcia procesu fotodegradacji. Bardzo często próbka jest utrwalana w celu regulacji odczynu roztworu, w zależności od kwasowego lub zasadowego charakteru badanej substancji. Próbka w miarę możliwości powinna być także w jak najkrótszym czasie poddana analizie, jeżeli jest to niemożliwe powinna być przechowywana w temperaturze od 4 oC do -20 oC [20, 26].
Z powodu bardzo niskich zawartości omawianych związków w próbkach środowiskowych jak i żywności, konieczna jest izolacja oraz znaczne wzbogacanie tych analitów z matrycy próbki, w celu osiągnięcia odpowiedniej granicy wykrywalności. Dlatego proces przygotowania próbki do analizy obejmuje kilka etapów, jakie schematycznie przedstawiono na schemacie (rys. 4.)
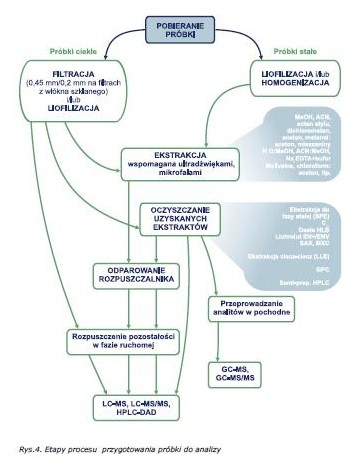
Procedury przygotowania próbek stałych do analizy takich jak gleba, osady denne, ściekowe lub tkanki zwierzęce są znacznie bardziej praco – i czasochłonne niż próbek ciekłych. Wyekstrahowanie analitów w tak niskich stężeniach z próbki stałej, zawierającej wiele substancji interferujących jest bardzo trudnym zadaniem [27-28].
Dokonując przeglądu literatury można zauważyć, iż najczęściej stosowaną metodą ekstrakcji antybiotyków z próbek środowiskowych jak i żywności jest technika ekstrakcji do fazy stałej SPE (ang. Solid Phase Extraction) [2-3, 17, 19, 23-24, 27-28]. Jest to metoda polegająca na przeniesieniu analitów z próbki ciekłej do fazy stałej – sorbentu, dzięki wykorzystaniu zjawiska podziału [26]. Na skutek silniejszego oddziaływania pomiędzy analitem a sorbentem analit ulega zaadsorbowaniu, a następnie wymywany jest z jego powierzchni odpowiednim rozpuszczalnikiem. Ekstrakcja do fazy stałej przeprowadzana jest najczęściej w odwróconym układzie faz przy zastosowaniu złoża w postaci żelu krzemionkowego modyfikowanego chemicznie grupami oktadecylowymi – C18 [29], aczkolwiek zastosowanie znajdują także inne sorbenty jak kopolimer diwinylobenzenu i winylopirolidonu (Oasis-HLB), wysokoporowate kopolimery o mieszanej polarności – etylowinylobenzen, diwinylobenzen, charakteryzujące się dużą powierzchnią aktywną i pojemnością (LiChrolut EN) [24, 30]. Sorbent Oasis-HLB nadaje się do ekstrakcji farmaceutyków o charakterze od kwaśnego po zasadowy w środowisku obojętnym, dzięki czemu można go zastosować w celu oznaczenia związków o różnym charakterze chemicznym w jednym cyklu analitycznym [31-32]. Przeprowadzono także badania mające na celu określenie stopnia odzysku analitów na różnych sorbentach [33]. Zaobserwowano, iż zastosowanie kationitu jako sorbentu sprzyja ekstrakcji związków o charakterze kwasowym, natomiast dla farmaceutyków obojętnych uzyskiwano bardzo niskie wartości odzysku. Isolut ENV+ znajduje zastosowanie w zatężaniu tylko niewielkiej liczby związków. Zalecany jest do związków polarnych w niskich wartościach pH. Natomiast odzyski uzyskane dla typowych wypełnień odwróconych (C18) charakteryzowały się dość wysokimi wartościami dla wszystkich badanych związków, aczkolwiek porównując je z złożem Oasis-HLB, były nieco niższe.
Technikę SPE cechuje prostota, wysoki odzysk analitów z matrycy, mniejsze zużycie toksycznych rozpuszczalników oraz zastosowanie mniejszych objętości próbki w porównaniu z ekstrakcją ciecz – ciecz LLE (ang. Liquid – Liquid Extraction). Bardzo ważną zaletą jest możliwość zastosowania różnorodnych ogólnodostępnych sorbentów, co przyczynia się do zwiększania efektywności tego procesu [34-35].
W literaturze proponowane jest także zastosowanie zautomatyzowanych metod ekstrakcji do fazy stałej przeznaczonych do oznaczania antybiotyków w próbkach środowiskowych takich jak mikroekstrakcja do fazy stałej SPME (ang. Solid Phase Microextraction). Wskazywane są także metody ekstrakcji wykorzystujące zjawiska powinowactwa immunochemicznego, których wyniki zatężania śladowych ilości farmaceutyków również mogłyby okazać się obiecujące.
Inną możliwą, coraz częściej wymienianą metodą ekstrakcji – jest zastosowanie tak zwanych polimerów z odciskami molekularnymi (z ang. Molecular Imprinted Polymers, MIP), które potrafią doskonale naśladować obiekty naturalne, takie jak enzymy i przeciwciała. Stąd wykorzystanie powinowactwa oznaczanych substancji do otrzymanych polimerów umożliwia ich selektywne wydzielenie. Polimery te mogą być zastosowane jako wypełnienie w kolumnach SPE lub mogą być upakowane w zwykłych kolumnach chromatograficznych [36]. Metoda ta jest także proponowana do oznaczania farmaceutyków w próbkach środowiskowych [27]. Złoża takie charakteryzują się bardzo dużą trwałością, są odporne na działanie silnych kwasów czy organicznych rozpuszczalników. Są bardzo specyficzne dla danego związku, co niestety stanowi ich wadę, bowiem niemożliwe jest wprowadzenie ich do rutynowych badań zawartości sumy zawartości leków w środowisku i żywności.
Chromatografia gazowa sprzęgnięta ze spektrometrią mas (GC-MS) lub tandemową spektrometrią mas (GC-MS/MS) znajduje również swoje zastosowanie do oznaczania pewnych grup farmaceutyków [27, 37], przy czym nie jest ona tak uniwersalna jak technika LC-MS z powodu zbyt wysokiej polarności tych analitów. Związki te, aby mogły być oznaczone z zastosowaniem chromatografii gazowej muszą być poddane procesowi derywatyzacji (spochodniania), w celu zwiększenia ich lotności jak i poprawy czułości metody. Jednakże z powodu obecności w ich budowie chemicznej bardzo różnorodnych grup funkcyjnych opracowanie uniwersalnej metody spochodniania wszystkich oznaczanych substancji pozostaje zadaniem bardzo trudnym. Dodatkowo, wprowadzenie dodatkowego procesu – przeprowadzania analitów w pochodne, powoduje, iż metoda staje się bardzo czasochłonna i pracochłonna. Ponadto, istnieje także możliwość zanieczyszczenia próbki lub jej częściowej utraty na skutek przeprowadzania dodatkowych operacji. Ponadto większość farmaceutyków jest termolabilna, więc ulegałaby degradacji w wysokiej temperaturze jaka panuje w chromatografie gazowym [27].
Między innymi z powyższych powodów techniki LC-MS i LC-MS/MS uważane są obecnie za najwłaściwsze metody oznaczania farmaceutyków w żywności i próbkach środowiskowych [5, 23]. Oprócz braku wymogu derywatyzacji oraz niskiej temperatury pracy aparatury techniki chromatografii cieczowej sprzęgniętej z tandemową spektrometrią mas pozwalają na rozdzielenie związków o tej samej masie cząsteczkowej, ale różniących się powstałymi jonami. Dodatkowo, możliwe jest jednoczesne oznaczenie związków należących do różnych grup kongenerycznych bez względu na ich polarność czy termolabilność [5, 38].
Analiza antybiotyków za pomocą tych metod polega na ich rozdzieleniu w odpowiednim układzie faz, a następnie detekcji za pomocą spektrometru mas. Anality są rozdzielane w kolumnie ze względu na różne powinowactwo do fazy stacjonarnej, a następnie eluowane za pomocą odpowiednio dobranych rozpuszczalników. Preferowane jest zastosowanie rozdzielania chromatograficznego w odwróconym układzie faz, stosując kolumny chromatograficzne z niepolarną fazą stacjonarną oraz polarne rozpuszczalniki i ich mieszaniny. Do tego celu najczęściej stosowane są mieszaniny metanol/woda, acetonitryl/woda przy odpowiednio dobranym pH z dodatkiem octanu amonu, mrówczanu amonu lub/i kwasu mrówkowego czy octowego, których dodatek powoduje zwiększenie czułości oznaczenia i poprawę jonizacji próbki [22, 24].
Identyfikacja rozdzielanych w kolumnie chromatograficznej antybiotyków na podstawie parametrów retencji jest na ogół metodą niewystarczającą, ponieważ w próbkach rzeczywistych istnieje wiele różnych substancji o charakterze interferetów, które mogą mieć bardzo zbliżony lub nawet identyczny czas retencji jak badane związki. Z tego względu, w celu dokonania poprawnej analizy jakościowej, produkty rozdzielania chromatograficznego są wraz z fazą ruchomą kierowane do spektrometru mas, gdzie następuje ich jonizacja i fragmentacja, natomiast identyfikacja poszczególnych związków potwierdzana jest na podstawie analizy ich widm mas [22, 30].
Możliwe jest zastosowanie chromatografii cieczowej sprzężonej ze spektrometrią mas z jonizacją na drodze elektrorozpraszania ESI (ang. Electrospray Ionization) lub z chemiczną jonizacją pod ciśnieniem atmosferycznym APCI (ang. Atmospheric Presure Chemical Ionisation). Jonizacja metodą elektrorozpraszania zaliczana jest do tak zwanych „miękkich” technik jonizacji próbki, prowadzących do otrzymania wyraźnego jonu molekularnego [5, 22]. W spektrometrii mas przedmiotem badań są jony powstałe na skutek jonizacji cząsteczki, rozdzielone w zależności od stosunku ich masy do ładunku (m/z) i następnie zarejestrowane, mierząc intensywność powstałego prądu jonowego. Najczęściej stosowanymi analizatorami mas jest pojedynczy (Q) lub potrójny (QqQ) kwadrupol, pułapka jonowa ITP. (ang. Ion Trap Detector) lub rzadziej analizator czasu przelotu TOF (ang. Time of Flight) [38, 39]. W wyniku jonizacji powstaje jon pseudomolekularny (w zależności od trybu pracy: [M-H]– dla ujemnej jonizacji lub [M+H]+w przypadku dodatniej jonizacji), odpowiadający masie cząsteczkowej substancji, który następnie ulega rozpadowi, dając inne jony fragmentacyjne. Powstałe w ten sposób jony są następnie rozdzielane w zależności od stosunku m/z w analizatorze, skąd przemieszczają się do detektora, który przekształca prąd jonowy w mierzalny sygnału.
Stosowane są różnego rodzaju detektory, takie, które dokonują bezpośredniego pomiaru ładunków docierających do detektora jak i zwiększające intensywność wykrywanego sygnału. Uzyskiwane ze spektrometru mas dane są przekształcane w zależności od potrzeb przez komputer w wykres zależności intensywności pików od wartości stosunku m/z. Bez względu na zastosowany sposób jonizacji próbki wyróżnia się trzy główne sposoby zbierania danych [40]:
- przemiatanie (ang. scanning) – w którym rejestruje się pełne widma mas między dwoma zadanymi skrajnymi wartościami mas;
- fragmentografię mas SIM (ang. selected ion monitoring) – stosuje się, gdy celem analizy jest wykrycie określonych substancji o znanych wcześniej widmach. Monitorowane są jedynie wybrane, charakterystyczne fragmenty jonów o określonych masach, które nie występują w potencjalnie interferujących innych związkach, a są wystarczające do identyfikacji danego antybiotyku. Jest to metoda, która odznacza się dużo większą czułością i selektywnością niż technika zbierania wszystkich jonów pochodzących z rozpadu danego związku chemicznego (total scanning);
- monitorowanie wybranych fragmentacji MRM (ang. Multiple Reaction Monitoring), w której sprawdza się obecność jonów fragmentacyjnych powstałych z danego jonu macierzystego, dzięki czemu przewyższa ona obie wyżej wymienione techniki zbierania danych i jest najbardziej miarodajną metodą potwierdzania obecności danego związku z próbce.
Oznaczanie farmaceutyków w próbkach środowiskowych przy zastosowaniu metody LC-MS lub LC-MS/MS pracującego w trybie SIM lub MRM charakteryzuje się wysoką czułością i selektywnością. W przypadku próbek środowiskowych o bardzo złożonym składzie matrycy jak próbki ścieków, osadów ściekowych czy tkanek zwierzęcych zalecane jest stosowanie właśnie tych trybów. Wadą tej techniki jest jednak niemożność zbierania i tworzenia biblioteki widm danych związków, jak to ma miejsce w przypadku analizowania związków metodą GC-MS. Widma mas uzyskiwane w metodzie LC-MS są bardzo zmienne, bardzo rzadko powtarzalne i zależą od wielu różnych czynników jak na przykład rodzaju fazy ruchomej, natężenie jej przepływu oraz zastosowane warunki analizy i jonizacji próbki.
Można przypuszczać, że dalsze badania w zakresie analityki farmaceutyków w tego typu próbkach będą skierowane przede wszystkim w stronę opracowania nowych procedur analitycznych umożliwiających oznaczenie znacznie większej liczby farmaceutyków, ich metabolitów na bardzo niskich poziomach stężeń w jednym cyklu analitycznym oraz na identyfikację i analizę ilościową produktów ich degradacji, stanowiących potencjalne zagrożenie dla organizmów żywych.
Uzyskane informacje będą stanowiły podstawę do wyciągnięcia odpowiednich wniosków jak i oceny trwałości danych farmaceutyków w środowisku, a to z kolei będzie przesłanką dla projektantów leków, którzy powinni tworzyć leki znacznie bardziej przyjazne dla środowiska, człowieka i zwierząt.
Zagrożenie jakie stwarza dziś obecność tych substancji w środowisku wobec człowieka i samego środowiska może ulec zmniejszeniu na skutek ograniczenia ich emisji poprzez prawidłowe i racjonalne stosowanie zarówno w produkcji żywności, hodowli i leczeniu zwierząt jak i medycynie człowieka.
Literatura
- R. Dobrzeniecka-Turek, W. Rusiniak, Chemia leków, Wydawnictwo Uczelniane Politechniki Szczecińskiej, Szczecin 1999.
- N. A. Botsoglou, D. J. Fletouris, Drug residues in foods – pharmacology, food safty and analysis, Mercel Dekker Inc., New York 2001.
- L. M. L. Nollet, Handbook of food analysis, Marcel Dekker Inc., New York 2004.
- N. Kemper, Veterinary antibiotics in the aquatic and terrestrial environment, Ecological Indicators, 8, 1-13, 2008.
- G. Balizs, A. Hewitt, Determination of veterinary drug residues by liquid chromatography and tandem mass spectrometry, Anal. Chim. Acta, 492, 105-131, 2003.
- T. Heberer, Occurrence, fate, and removal of pharmaceuticals residues in the aquatic environment: a review of recent research data, Toxicol. Lett., 131, 5-17, 2002.
- B. Halling-Sorensen i współpr., Occurrence, fate and effects of pharmaceutical substances in the environment – A review, Chemosphere, 36, 357-393, 1998.
- K. Kummerer, Pharmaceuticals in the environment-sources, fate, effects and risks, Springer, Berlin, Heidelberg 2004.
- K. Sarmah, M. T. Meyer, A. B. A. Boxall, A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment, Chemosphere, 65, 725-759, 2006.
- T. Reemtsma, M. Jekel, Organic pollutants in the water cycle, WILEY-VCH, Weinheim 2006.
- F. Sacher, F. T. Lange, H. Brauch, I. Blankenhom, Pharmaceuticals in ground waters. Analytical methods and result of a monitoring program in Baden-Wurttemberg, Germany, J. Chromatogr. A, 938, 199-210, 2001.
- M. Clara i współpr., Removal of selected pharmaceuticals, fragrances and endocrine disrupting compound in a membrane bioreactor and conventional wastewater treatment plants, Water Res., 39, 4797-4807, 2005.
- Y. Yoon et al., Removal of endocrine disrupting compounds and pharmaceuticals by nanofiltration and ultrafiltration membranes, Desalination, 202, 16-23, 2007.
- A. Gulkowska, H.W. Leung i współpr., Removal of antibiotics from wastewater by sewage treatment facilities in Hong Kong and Shenzhen, China, Water Res., Article in Press, 2007.
- J. Watkinson, E. J. Murby, S. D. Costanzo, Removal of antibiotics in conventional and advanced wastewater treatment: Implications for environmental discharge and wastewater recycling, Water Res., 41, 4164-4176, 2007.
- T. I. Jones-Lepp, R. Stevens, Pharmaceuticals and personal care products in biosolids/sewage sludge: the interface between analytical chemistry and regulation, Anal. Bioanal. Chem., 387, 1173-1183, 2007.
- T. A. Ternes, Analytical methods for the determination of pharmaceuticals in aqueous environmental samples, Trends Anal. Chem., 20, 419-434, 2001.
- F. Ingerslev, i współpr., Primary biodegradation of veterinary antibiotics in aerobic and anaerobic surface water simulation system, Chemosphere, 44, 865-872, 2001.
- J. Dębska, A. Kot-Wasik, J. Namieśnik, Pozostałości środków farmaceutycznych w środowisku – przemiany, stężenia, oznaczanie, Chem. Inż. Ekol., 8, 723-750, 2003.
- A. Kot-Wasik, J. Dębska, J. Namieśnik, Analytical techniques in studies of the environmental fate of pharmaceuticals and personal care products, Trends Anal. Chem., 26, 557-568, 2007.
- J. Dębska, Oznaczanie pozostałości farmaceutyków o charakterze kwasowym, zasadowym i neutralnym w próbkach środowiskowych, Praca Doktorska, Wydział Chemiczny, Politechnika Gdańska, Gdańsk 2006.
- W. M. A. Niessen, Analysis of antibiotics by liquid chromatography–mass spectrometry, J. Chromatogr. A, 812, 53-75, 1998.
- P. Munoz i współpr., A versatile liquid chromatography-tandem mass spectrometry system for the analysis of different groups of veterinary drugs, Anal. Chim. Acta, 529, 137-144, 2005.
- M. J. López de Alda, S. Diaz-Cruz, M. Petrović, D. Barceló, Liquid chromatography – (tandem) mass spectrometry of selected emerging pollutants (steroid sex, drugs and alkyphenolic surfactants) in the aquatic environment, J. Chromatogr. A, 1000, 503-526, 2003.
- E. P. Popek, Sampling and analysis of environmental chemical pollutants, Academic Press, Walnut Creek 2003.
- J. Namieśnik, Z. Jamrógiewicz, M. Pilarczyk, L. Torres, Przygotowanie próbek środowiskowych do analizy, WNT, Warszawa 2000.
- M. S. Diaz-Cruz, J. López de Alda, D. Barceló, Environmental behavior and analysis of veterinary and human drugs in soils, sediments and sludge, Trends Anal. Chem., 22, 340-351, 2003.
- D. Mutavdźić Pavlović, S. Babić, A. J. M. Horvat, M. Kastelan-Macan, Sample preparation in analysis of pharmaceuticals, Tredns Anal. Chem., 26, 1062-1074, 2007.
- J. Zhu i współpr., Analysis of oxytetracycline, tetracycline and chlorotetracycline in water using solid-phase extraction and liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 928, 177-186, 2001.
- M. J. Gomez, M. Petrović, A. R. Fernandez-Alba, D. Barceló, Determination of pharmaceuticals of various therapeutic classes by solid – phase extraction and liquid chromatography – tandem mass spectrometry analysis in hospitals effluent wastewater, J. Chromatogr. A, 1114, 224-233, 2006.
- W. W. Buchberger, Novel analytical procedures for screening of drugs residues in water, waste water, sediment and sludge, Anal. Chim. Acta, 593, 129-139, 2007.
- S. Ollers i współpr., Simultaneous quantification of neutral and acidic pharmaceuticals and pesticides at the low – ng/l level in surface and waste water, J. Chromatogr. A, 911, 225-234, 2001.
- M. Gros, M. Petrović, D. Barceló, Development of multi-residue analytical methodology based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) for screening and trace level determination of pharmaceuticals in surface and wastewaters, Talanta, 70, 678-690, 2006.
- W. Kleibohmer, Handbook of analytical separations, Elsevier, 3, 310-319, 2001.
- M. C. Hennion, Solid-phase extraction: method development, sorbents and coupling with liquid chromatography, J. Chromatogr. A, 856, 3-54,1999.
- C. He i współpr. , Application of molecularly imprinted polymers to solid-phase extraction of analytes from real samples, J. Biochem. Biophsys. Methods, 70, 133-150, 2007.
- W. C. Lin, H. C. Chen, W. H. Ding, Determination of pharmaceuticals residues in waters by solid-phase extraction and large-volume on-line derivatization with gas chromatography – mass spectrometry, J. Chromatogr. A, 1065, 279-285, 2005.
- D. Fatta, A. Nikolaou, A. Achilles, S. Mric, Analytical methods for tracing pharmaceuticals residues in water and wastewater, Trends Anal. Chem., 26, 515-533, 2007.
- S. Perez, D. Barceló, Application of advanced MS techniques to analysis and identification of human and microbial metabolites of pharmaceuticals in the aquatic environment, Trends Anal. Chem., 26, 494-514, 2007.
- E. de Hoffmann, J. Charette, V. Stroobant, Spektrometria mas, WNT, Warszawa 1998.
*Katedra Analizy Środowiska, Wydział Chemii Uniwersytetu Gdańskiego