Matryca paszowa i przygotowanie próbki a jakość wyników badań składników analitycznych pasz
Autor: Waldemar Korol, Grażyna Bielecka, Jolanta Rubaj, Sławomir Walczyński*
Streszczenie
Celem pracy była ocena wpływu niejednorodności (homogeniczności) matrycy paszowej i przygotowania próbki do badań z próbki laboratoryjnej na miarodajność wyników badań i niepewności pomiarów składników analitycznych pasz i dodatków paszowych. Zakres badań obejmował wybrane składniki odżywcze (białko, popiół), makroelementy (Ca, P, Na, Cl), mikroelementowe dodatki paszowe (Fe, Mn, Zn, Cu, Se, J, Mo) i dodatki paszowe dietetyczne (lizyna, metionina, witamina A). Podano sposób obliczenia zmienności technicznej badanego składnika w próbce do badań, wydzielonej z próbki laboratoryjnej, jako miary niehomogeniczności i niepewności przygotowania próbki do badań. Potwierdzono, że właściwe wydzielenie próbki do badań z próbki laboratoryjnej i jej rozdrobnienie poprawiło precyzję badań i obniżyło niepewność pomiaru a średnie wyniki badań składników w poprawnie przygotowanych próbkach były w większym stopniu zgodne z deklaracjami na etykietach produktów.
Wstęp
Przygotowanie próbki paszy do badań może być głównym źródłem błędu laboratoryjnego, co jest zwykle niedostatecznie dostrzegane. Błąd popełniony na etapie przygotowania próbki w laboratorium może wpływać na wynik badania w większym stopniu niż błąd w badaniach analitycznych [16].
Pasze są z natury materiałem niejednorodnym, którego heterogeniczność naturalna powoduje, że nie wszystkie cząstki próbki laboratoryjnej posiadają ten sam skład i różnią się kształtem, wielkością, gęstością. Ten rodzaj heterogeniczności, zwany heterogenicznością składu lub naturalną, nie zależy od stopnia wymieszania. Można ją zmniejszyć poprzez rozdrabnianie i redukcję średniej wielkości cząstek. Dlatego w celu zmniejszenia heterogeniczności naturalnej konieczne jest czasami wstępne rozdrabnianie całej próbki laboratoryjnej o wielkości cząstek > 6 mm, przed podziałem i przygotowaniem próbki do badań, zwykle o masie około 100 g, która następnie jest rozdrabniana do wielkości cząstek zależnej od wykonywanych badań. Błąd przygotowania próbki analitycznej związany z heterogenicznością składu może być ograniczany dzięki wybraniu odpowiedniej masy odważki analitycznej do badań [16].
Innym rodzajem niejednorodności próbki paszy jest heterogeniczność rozkładu, która jest miarą nielosowej dystrybucji cząstek w próbce, wynikającej głównie z działania siły grawitacji na cząstki o różnych gęstościach, wielkościach i kształtach, co prowadzi do grupowania i segregacji cząstek a nawet do rozwarstwiania, przy czym cząstki najmniejsze lub o największej gęstości opadają na dno próbki. W celu zmniejszenia błędu grupowania i segregacji cząstek (heterogeniczności rozkładu), należy wymieszać lub zmiksować próbkę przed przygotowaniem próbki do badań i zebrać wiele losowych części z próbki laboratoryjnej [16]. W przypadku niektórych produktów paszowych, takich jak sypkie mieszanki paszowe uzupełniające MPU o dużej zawartości składników mineralnych i zróżnicowanej gęstości, niewłaściwe mieszanie może zwiększyć segregację, zamiast ją zredukować.
Inne błędy, które wynikają z przygotowania próbki, w tym utrata lub przyrost zawartości analitu, związane są z rozdrabnianiem, utratą drobnych cząstek, zanieczyszczeniem i segregacją elektrostatyczną. Przyczyną błędów może być lotność oznaczanych substancji w związku z nagrzewaniem próbki na skutek tarcia podczas rozdrabniania. Błędy te mogą być duże i są zwykle wynikiem nieostrożności lub braku wiedzy. W Tabeli 1 przedstawiono klasyfikację parametrów stabilnych i niestabilnych oraz przyczyn degradacji w odniesieniu do przygotowania próbki paszy – wg PN EN-ISO 6498 [16]. W przypadku badania parametrów stabilnych zaleca się rozdrabnianie próbki do średniej wielkości cząstek 0,5 mm lub niższej, natomiast w przypadku parametrów niestabilnych, aby ograniczyć wzrost temperatury, zaleca się rozdrabnianie próbki do średniej wielkości cząstek 1 mm.
Tabela 1. Klasyfikacja parametrów stabilnych i niestabilnych oraz przyczyn degradacji w odniesieniu do przygotowania próbki paszy – wg PN EN-ISO 6498 [16]
|
Pochodzenie |
Parametry stabilne |
Parametry niestabilne |
Przyczyna degradacji/zmiany |
|
Składniki odżywcze |
(Surowe) białko, tłuszcz, popiół, włókno |
Wilgoć |
Temperatura (lotność) |
|
Skrobia, cukier, laktoza |
Amoniak |
Temperatura (lotność) |
|
|
Wytwarzanie gazu i wytwarzanie rozpuszczalnej substancji organicznej z użyciem enzymu w testach in vitro |
Kwasy organiczne (np. kwas mlekowy, kwas octowy, kwas masłowy, kwas fumarowy, kwas mrówkowy) |
Temperatura (lotność) |
|
|
Składniki mineralne (np. Ca, P, Mg, Na, K, Cl) |
Nienasycone kwasy tłuszczowe |
Utlenianie w powietrzu (może skutkować wytwarzaniem krótkołańcuchowych kwasów tłuszczowych) |
|
|
Dodatki paszowe |
Pierwiastki śladowe |
Witaminy |
Temperatura, światło ultrafioletowe (UV) utlenianie w powietrzu (substancje wrażliwe) |
|
Aminokwasy (np. lizyna, metionina, tryptofan) |
1,2-propanodiol, glikol etylenowy |
Temperatura (lotność) |
|
|
Enzymy (np. fitazy, enzymy rozkładające polisacharydy nieskrobiowe) |
Mikroorganizmy jak probiotyki (np. Saccharomyces cerevisiae, Enterococcus faecium) |
Temperatura (zamrażanie), ciśnienie (substancje wrażliwe na rozdrabnianie); wilgoć/suchość (wpływa na wzrost mikroorganizmów) |
|
|
Substancje niepożądane |
Metale ciężkie |
Mikotoksyny (np. aflatoksyna B1, deoksyniwalenol, fumonizyny, ochratoksyna A, toksyna T-2, toksyna HT-2, zearalenon, alkaloidy sporyszu) |
Wzrost pleśni i zmiana mikotoksyn możliwa w temperaturze pokojowej; światło UV (substancja wrażliwa – aflatoksyna B1) |
|
Dioksyny i dioksynopodobne polichlorowane bifenyle (PCB) |
Leki, antybiotyki, pestycydy |
Temperatura (substancje wrażliwe) |
|
|
|
Kwas cyjanowodorowy |
Temperatura (lotność) |
|
|
Substancje zakazane |
Białka pochodzenia zwierzęcego |
Zakazane leki, zakazane antybiotyki |
Temperatura (substancje wrażliwe) |
|
Inne – mikro-organizmy |
|
Drożdże, bakterie, pleśnie |
Temperatura (substancje wrażliwe), suszenie, dopływ tlenu (anaerobioza) |
Zgodnie z aktualnymi wymaganiami w zakresie badania pasz, wynik badania przedstawiany jest jako X±U, gdzie X jest wartością średnią a U niepewnością rozszerzoną pomiaru dla współczynnika rozszerzenia k=2. Taki sposób przedstawiania wyniku jest niezbędny, np. do stwierdzenia zgodności z wymaganiami [4, 18, 19, 20]. Wynik pomiaru powinien być miarodajny (ważny), podobnie jak niepewność pomiaru, a wymagania dotyczące stwierdzeń zgodności podano w p. 7.8.6 normy PN-EN ISO/IEC 17025 [14].
W związku z ryzykiem popełnienia błędu przygotowania próbki w przypadku matryc paszowych charakteryzujących się niejednorodnością, celem niniejszej pracy było podanie przykładów postępowania, w zależności od rodzaju matrycy paszowej, ograniczającego ryzyko błędu przygotowania próbki do akceptowanego poziomu i ocena wpływ postępowania na wyniki i niepewności pomiaru badanych składników [9, 10, 11].
Materiały i metody
Materiałem do oceny homogeniczności próbek laboratoryjnych były granulowane mieszanki paszowe pełnoporcjowe MPP, sypkie mieszanki paszowe uzupełniające MPU oraz premiksy paszowe (przedmieszki mineralno-witaminowe).
Homogeniczność próbki laboratoryjnej obliczono wykorzystując prawo propagacji błędów Gauss’a. Istotą proponowanego postępowania jest rozdzielenie dwóch składowych zmienności wyniku pomiaru homogeniczności wyrażonej współczynnikiem zmienności CVh+a (%): zmienności analitycznej CVa (%) i zmienności technicznej CVh (%) odpowiadającej niejednorodności (niehomogeniczności) badanego składnika podczas przygotowania próbki do badań w laboratorium. Zmienność analityczną metody obliczano z rozstępu, na przykładzie Raportu Technicznego Nordtest [12]. Do obliczeń wykorzystano poniższe wzory, (1) i (2).
(1) 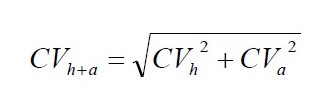
(2) 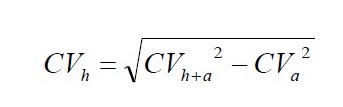
Przyjęto, że współczynnik zmienności technicznej CVh jako parametr oceny homogeniczności próbki laboratoryjnej jest miarą niepewności przygotowania próbki i może być uznany jako standardowa niepewność przygotowania próbki (CVh = uh/√n; gdzie n = liczna próbek do badań wyodrębnionych z próbki laboratoryjnej) w budżecie niepewności pomiaru uh+a uwzględniającym niepewność przygotowania próbki laboratoryjnej i niepewność postępowania analitycznego i pomiaru, obliczonym wg wzoru (3),
(3) 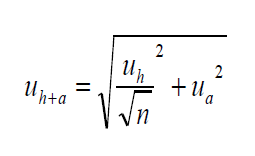
w którym ua jest niepewnością postępowania analitycznego i pomiaru. Niepewność rozszerzoną Uh+a dla współczynnika rozszerzenia k=2 (P=95%) obliczano jak następuje: Uh+a = uh+a · 2.
Obliczono niepewności przygotowania próbki do badań wybranych podstawowych składników pokarmowych (białko, popiół), makroelementów (wapń, sód, chlorki, fosfor), mikroelementowych dodatków paszowych (żelazo, mangan, cynk, miedź, selen, jod, molibden) oraz dodatków paszowych dietetycznych (witamina A, lizyna, metionina).
Do oceny homogeniczności i niepewności przygotowania próbki laboratoryjnej, pobierano próbki laboratoryjne pasz o masie około 0,5 kg zgodnie z rozporządzeniem 691/2013 [17]. Z każdej próbki laboratoryjnej przygotowywano, wg zaleceń normy [16], 6 próbek do badań o masie około 100 g każda, stosując rozdzielacz obrotowy. W przypadku sypkich MPU i granulowanych MPP, próbki do badań rozdrabniano w całości z przesiewem przez sito 0,5 mm. W przypadku MPU DJ 2% próbki dodatkowo rozdrabniano w młynie planetarnym do wielkości cząstek 0,25 mm. W przypadku badania homogeniczności witaminy A w MPP i MPU, próbki laboratoryjne po rozdzieleniu rozdrabniano w rozdrabniaczu z przesiewem przez sito 1 mm. Próbki premiksu badano bez rozdrabniania (potwierdzona homogeniczność) i po rozdrobnieniu w młynie kulowym do wielkości cząstek około 0,25 mm. W tak przygotowanych próbkach wykonywano oznaczenia zawartości wybranych składników pokarmowych (białko, popiół) i składników mineralnych (Ca, Na, Cl–, Zn, Mn, Cu, Mo, Se, J) oraz pozostałych dodatków w dwóch powtórzeniach.
Zawartości białka, popiołu, witaminy A, lizyny i metioniny badano metodami oficjalnymi, zgodnie z rozporządzeniem 152/2009 [18]. Zawartości wapnia, sodu, cynku i manganu oznaczono metodą płomieniowej absorpcyjnej spektrometrii atomowej FAAS [13]. Chlorki oznaczono metodą biamperometrycznego miareczkowania [8]. Selen badano metodą generowania wodorków HGAAS [2] a molibden metodą spektrometrii mas z indukcyjnie sprzężoną plazmą ICP-MS [3]. Również jod badano metodą ICP-MS [15]. Średnią wielkość cząstek premiksu przed rozdrobnieniem i po rozdrobnieniu mierzono optyczno-elektronicznym miernikiem wielkości i kształtu cząstek AWK 3D.
Z równania (2) obliczono współczynnik zmienności technicznej (niehomogeniczności) próbki laboratoryjnej na podstawie wyników badań próbek wydzielonych z próbki laboratoryjnej. Z równania (3) obliczono niepewności przygotowania próbki do badań w laboratorium i niepewności postępowania analitycznego i pomiaru.
Sposób obliczania zmienności technicznej CVh i niepewności przygotowania próbki laboratoryjnej na przykładzie witaminy A w sypkiej MPU podano w Tabeli 2.
Tabela 2. Obliczanie zmienności technicznej CVh (homogeniczności) próbki laboratoryjnej i niepewności przygotowania próbki na przykładzie badania witaminy A w sypkiej MPU
|
Wyszczególnienie |
Sposób obliczenia – wzory |
|
Współczynnik zmienności (n=12) CVh+a = 12,7% |
Dane z laboratorium – szablon Excel |
|
Powtarzalność obliczona z rozstępu wg [12], (n=6) CVa = 3,66% |
Dane z laboratorium – szablon Excel |
|
Współczynnik zmienności technicznej i niepewność przygotowania próbki CVh = 11,8% = uh |
Wzór: |
|
Standardowa niepewność badania witaminy A z budżetu niepewności u = 9,7%; U=19,4% |
Dane z walidacji metody |
|
Standardowa niepewność badania wit. A z niepewnością przygotowania próbki w laboratorium uh+a = 15,3% (n=1); uh+a = 10,85% (n=6); |
Wzór:
|
|
Niepewność rozszerzona Uh+a = 30,6% (n=1); Uh+a = 21,7% (n=6); |
15,3% x 2 = 30,6% (n=1) 10,85% x 2 = 21,7% (n=6) |
Wyniki i dyskusja
Podany przykład obliczania homogeniczności próbki laboratoryjnej i niepewności przygotowania próbki do badań, oparty na rzeczywistych wynikach badania MPU wskazuje, że ta składowa niepewności pomiaru może mieć istotny udział w budżecie niepewności (Tabela 2). Współczynnik zmienności CVh witaminy A w próbkach do badań wyodrębnionych z sypkiej MPU był wysoki i wyniósł 11,8%. Niepewność rozszerzona, obliczona wg GUM [5], która na etapie walidacji metody została określona na poziomie 19,4%, wzrosła do 30,6% (dla n=1) po uwzględnieniu niepewności przygotowania próbki do badań w laboratorium. Na wysoką wartość niepewności wpłynęły cechy morfologiczne zastosowanego preparatu witaminy A, związane z otoczkowaniem substancji czynnej w celu zabezpieczenia przed utlenianiem. Podobną niepewność rozszerzoną pomiaru witaminy A w mieszankach paszowych, w tym w MPU, wynoszącą 30%, stosują niemieckie laboratoria VDLUFA upoważnione do badań pasz w ramach urzędowego nadzoru [21].
W Tabeli 3 podano wyniki badania zmienności technicznej CVh (%) próbki laboratoryjnej i niepewności pomiaru uwzględniającej niepewność przygotowania próbki dla wybranych składników pokarmowych i dodatków paszowych w mieszance paszowej sypkiej i granulowanej. Oszacowane niepewności wyników pomiaru białka, popiołu oraz składników mineralnych z uwzględnieniem niepewności przygotowania próbki Uh+a w przypadku sypkiej MPU okazały się wyższe, od 20% do 80%, średnio o 48%, od niepewności U oszacowanej bez uwzględniania składowej przygotowania próbki (dla n=1). Przyczyną istotnego wpływu przygotowania próbki na wyniki szacowania niepewności w sypkiej MPU była heterogeniczność próbki laboratoryjnej i skłonność do segregacji składników, głównie składników mineralnych i witaminy A. W przypadku mikroelementów, selenu i molibdenu, wpływ na wysoką zmienność techniczną CVh i niepewność pomiaru mogła mieć także niska odważka analityczna (0,5 g), wynikająca z zastosowania mineralizacji mikrofalowej do rozkładu materiału.
W przypadku granulowanej MPP, wpływ składowej niepewności przygotowania próbki na całkowitą niepewność pomiaru był mniejszy. Uwzględnienie niepewności przygotowania próbki w niepewności pomiaru spowodowało wzrost oszacowanej niepewności od 2% do 20%, średnio o 8%. W rezultacie dla mieszanki granulowanej uzyskano podobne wyniki szacowania niepewności, świadczące o poprawie homogeniczności granulowanego produktu przez ograniczenie segregacji składników (Tabela 3).
Tabela 3. Wyniki badania zmienności technicznej CVh próbki laboratoryjnej i niepewności pomiaru z uwzględnieniem niepewności przygotowania próbki w sypkiej MPU i granulowanej MPP
|
Składnik paszy |
m, g
|
MPU – sypka |
MPP – granulowana |
||||||||
|
X
|
CVh % |
U % |
UH+A (n=1) % |
UH+A (n=6) % |
X |
CVh % |
U % |
UH+A (n=1) % |
UH+A (n=6) % |
||
|
Białko, g/kg |
0,5 |
288 |
1,53 |
3,0 |
4,3 |
3,25 |
196 |
1,33 |
4,0 |
4,8 |
4,14 |
|
Popiół, g/kg |
5 |
362 |
2,67 |
4,0 |
6,7 |
4,55 |
46,9 |
0,98 |
4,2 |
4,6 |
4,27 |
|
Wapń, g/kg |
5 |
125 |
3,59 |
9,6 |
12,0 |
10,0 |
8,36 |
1,47 |
9,6 |
10,0 |
9,67 |
|
Sód, g/kg |
5 |
4,71 |
4,65 |
11,6 |
14,9 |
12,2 |
1,50 |
1,53 |
11,6 |
12,0 |
11,7 |
|
Chlorki, g/kg |
2 |
6,59 |
3,45 |
8,4 |
10,9 |
8,85 |
3,16 |
0,92 |
9,7 |
9,9 |
9,73 |
|
Cynk, mg/kg |
5 |
233 |
5,02 |
10,4 |
14,5 |
11,2 |
198 |
2,52 |
10,4 |
11,6 |
10,6 |
|
Selen, mg/kg |
0,5 |
0,89 |
11,5 |
15,0 |
27,4 |
17,7 |
0,28 |
3,55 |
15,0 |
16,6 |
15,3 |
|
Molibden, mg/kg |
0,5 |
2,40 |
11,5 |
20,0 |
30,5 |
22,1 |
1,86 |
5,29 |
20,0 |
22,6 |
20,5 |
|
Wit. A*, mg/kg |
20 |
27,0 |
11,8 |
19,4 |
30,6 |
21,7 |
2,61 |
3,39 |
20,0 |
21,1 |
20,2 |
* w przeliczeniu na retinol (90000 j.m./kg); m – odważka analityczna w gramach; X – zawartość; U – niepewność rozszerzona (k=2) wg GUM; CVh – współczynnik zmienności technicznej; UH+A – niepewność rozszerzona (k=2) z niepewnością przygotowania próbki (n=1; n=6)
Stopień rozdrobnienia próbki może wpływać na wyniki badań [16]. Wpływ rozdrobnienia premiksu na wyniki oceny zmienności technicznej CVh (homogeniczności) próbki laboratoryjnej i niepewność pomiaru podano w Tabeli 4. Rozdrobnienie premiksu w młynie kulowym i prawie dwukrotne zmniejszenie średnicy cząstek spowodowało trzykrotne obniżenie zmienności technicznej CVh i poprawę homogeniczności, średnio z 7,76% w przypadku premiksu nie rozdrobnionego do 2,40% w premiksie rozdrobnionym. Należy podkreślić, że dla premiksu nierozdrobnionego potwierdzono homogeniczność na podstawie badania zawartości chlorków (masa odważki analitycznej 2 g), stosując uznaną procedurę badania homogeniczności przyjętą w badaniach biegłości [7]. Rozdrobnienie premiksu wpłynęło na obniżenie niepewności pomiaru, w przypadku cynku i miedzi prawie dwukrotnie, zwłaszcza przy badaniu jednej, wydzielonej próbki do badań (n=1), co jest często praktykowane w laboratoriach. Wyniki badań potwierdziły możliwość istotnego obniżenia niepewności pomiaru przez zwiększenie ilości próbek do badań pobranych z próbki laboratoryjnej (od n=1 do n=6) i poddanie ich analizie ale łatwiejszym, prostszym rozwiązaniem jest rozdrobnienie próbki do mniejszych rozmiarów cząstek. Prezentowane w Tabeli 4 niepewności pomiaru składników premiksu badanego bez rozdrabniania i obliczone na podstawie badania sześciu wydzielonych próbek do badań były wyższe od niepewności pomiaru składników w rozdrobnionym premiksie w jednej próbce do badań. Należy ocenić, czy rozdrabnianie nie wpływa na stabilność analitu, korzystając z wytycznych do przygotowania próbki [16].
Prezentowane dane wskazywały, że zależna od stopnia rozdrobnienia większa niejednorodność (heterogeniczność) próbki laboratoryjnej i wyższy współczynnik CVh wpływała przede wszystkim na niepewność pomiaru, zależnie od ilości próbek do badań wydzielonych z próbki laboratoryjnej. Nie stwierdzono wyraźnego wpływu stopnia rozdrabniania na wartości średnie, z wyjątkiem cynku (Tabela 4).
Tabela 4. Wyniki badania zmienności technicznej CVh (%) próbki laboratoryjnej i niepewności pomiaru uwzględniające niepewność przygotowania próbki laboratoryjnej wybranych składników mineralnych premiksów
|
Składnik premiksu |
m, g |
Premiks bez rozdrabniania*, średnica cząstek 416 μm |
Premiks rozdrobniony*, średnica cząstek 260 μm |
||||||||
|
X |
U % |
CVh % |
UH+A (n=1) % |
UH+A (n=6) % |
X |
U % |
CVh % |
UH+A (n=1) % |
UH+A (n=6) % |
||
|
Wapń, g/kg |
1 |
175 |
7,4 |
5,77 |
13,7 |
8,77 |
170 |
7,4 |
0,70 |
7,6 |
7,42 |
|
Żelazo, g/kg |
1 |
13,2 |
9,4 |
5,76 |
14,9 |
10,5 |
13,2 |
9,4 |
1,67 |
10,0 |
9,50 |
|
Mangan, g/kg |
1 |
14,2 |
10,4 |
7,07 |
17,6 |
11,9 |
14,0 |
10,4 |
2,56 |
11,6 |
10,6 |
|
Cynk, g/kg |
1 |
9,22 |
10,0 |
10,1 |
22,5 |
13,0 |
9,85 |
10,0 |
3,88 |
12,7 |
10,5 |
|
Miedź, g/kg |
1 |
1,42 |
10,6 |
10,1 |
22,8 |
13,4 |
1,44 |
10,6 |
3,17 |
12,3 |
10,7 |
m – masa odważki analitycznej w gramach; X – wartość średnia; U – niepewność rozszerzona (k=2) wg GUM [5], obliczona bez uwzględniania niepewności przygotowania próbki; CVh – homogeniczność – współczynnik zmienności technicznej; UH+A – niepewność rozszerzona (k=2) z niepewnością przygotowania próbki (n=1; n=6); * homogeniczność premiksu potwierdzona w badaniu chlorków zgodnie z normą ISO 13528 [7]
Na podstawie wyników badań prezentowanych w Tabeli 5 stwierdzono, że właściwe przygotowanie próbek do badań MPU (rozdrobnienie do wielkości cząstek 260 μm), czterokrotnie poprawiło precyzję badań a średnie wyniki badań rozdrobnionych próbek MPU były w większym stopniu zgodne z deklaracjami na etykiecie. Rozdrabnianie MPU o wysokiej zawartości składników mineralnych ponad 700 g/kg (MPU DJ 2% – udział w mieszance paszowej na poziomie 2%) do wielkości cząstek około 0,25 mm w badaniach składników mineralnych jest niezbędne, podobnie jak w przypadku premiksów, zwłaszcza przy niewielkich odważkach analitycznych, niższych od 1 g [1].
Należy podkreślić, że podobnie jak w przypadku premiksu (Tabela 4), rozdrobnienie próbki wpłynęło w większym stopniu na niepewności pomiarów niż na wartości średnie. W przypadku składników mineralnych badanej MPU (DJ 2%), miedzi, żelaza, wapnia i selenu, w nierozdrobnionej próbce odnotowano zmienność analityczną CVa większą niż zmienność całkowita CVh+a co uniemożliwiło obliczenie zmienności technicznej (ujemna wartość pod pierwiastkiem) będącej miarą homogeniczności CVh (Tabela 5). Po rozdrobnieniu próbki nie stwierdzono żadnego przypadku, w którym zmienność całkowita byłaby mniejsza od zmienności analitycznej. Jest to kolejny dowód na istotny wpływ stopnia rozdrobnienia próbki do badań, zwłaszcza w przypadku MPU, na miarodajność (ważność) wyników badań i niepewności pomiarów.
Tabela 5. Wyniki badania zawartości składników pokarmowych i dodatków paszowych w MPU- DJ 2% w próbkach nierozdrobnionych i rozdrobnionych (n=8) oraz wpływ stopnia rozdrobnienia na zmienność wyników i ocenę zgodności z deklaracją na etykiecie [1]
|
Składnik paszy |
D |
m, g |
u % |
MPU-DJ 2% – nierozdrobniona, około 0,5 mm |
MPU-DJ 2% – rozdrobniona, około 0,25 mm |
||||||
|
X |
CVh+a % |
CVh, % |
uhn % |
X |
CVh+a, % |
CVh, % |
uhr % |
||||
|
Białko, g/kg |
62,7 |
0,5 |
2,5 |
72,5 |
13,04 |
13,0 |
9,5 |
67,6 |
2,24 |
2,22 |
2,9 |
|
Popiół, g/kg |
820 |
5 |
3,0 |
751 |
0,91 |
0,89 |
3,1 |
734 |
0,66 |
0,66 |
3,0 |
|
Lizyna, g/kg |
12,5 |
0,5 |
5,0 |
14,5 |
7,38 |
5,36 |
6,3 |
13,9 |
5,19 |
2,44 |
5,3 |
|
Metionina, g/kg |
63 |
0,5 |
7,5 |
69,6 |
9,37 |
7,48 |
9,2 |
59,8 |
2,82 |
1,97 |
7,6 |
|
Cynk, mg/kg |
3000 |
1 |
5,0 |
2500 |
20,5 |
6,93 |
7,0 |
2920 |
2,36 |
0,88 |
5,0 |
|
Miedź, mg/kg |
400 |
1 |
7,5 |
458 |
20,7 |
-* |
-* |
385 |
3,98 |
2,66 |
7,7 |
|
Mangan, mg/kg |
4250 |
1 |
5,0 |
4040 |
20,1 |
9,37 |
8,3 |
4180 |
1,96 |
1,39 |
5,1 |
|
Żelazo, mg/kg |
2250 |
1 |
5,0 |
3090 |
20,3 |
-* |
-* |
2800 |
3,23 |
2,33 |
5,3 |
|
Wapń, g/kg |
200 |
1 |
5,0 |
180 |
5,68 |
-* |
-* |
193 |
1,39 |
1,05 |
5,1 |
|
Sód, g/kg |
71 |
1 |
5,0 |
68,6 |
11,1 |
3,94 |
5,7 |
72,7 |
1,51 |
0,27 |
5,1 |
|
Fosfor, g/kg |
51,2 |
1 |
3,1 |
43,6 |
4,59 |
2,57 |
4,6 |
50,7 |
1,01 |
0,89 |
3,2 |
|
Jod, mg/kg |
50 |
0,3 |
11,5 |
40,7 |
107 |
36,7 |
28,4 |
49,7 |
12,5 |
12,0 |
14,3 |
|
Selen, mg/kg |
12,5 |
0,5 |
7,5 |
20,7 |
78,3 |
-* |
-* |
11,8 |
21,7 |
7,74 |
9,3 |
|
Średnia |
|
|
5,4 |
|
23,2 |
9,6 |
9,1 |
|
4,56 |
2,76 |
5,8 |
D – deklaracja na etykiecie; masa odważki analitycznej w gramach; CVh+a – współczynniki zmienności wyników; CVh – współczynniki zmienności technicznej; * zmienność całkowita CVh+a mniejsza od zmienności analitycznej CVa obliczonej wg Nordtest [12] – niemożliwe obliczenie zmienności technicznej; u – standardowa niepewność złożona obliczona wg GUM [5] na etapie sprawdzania metod; uhn – standardowa niepewność złożona dla próbek nierozdrobnionych z uwzględnieniem niepewności przygotowania próbki (dodano składową CVh /√2); uhr – standardowa niepewność złożona dla próbek rozdrobnionych z uwzględnieniem niepewności przygotowania próbki (dodano składową CVh /√2)
Podsumowanie
Niejednorodność (niehomogeniczność) próbki laboratoryjnej powinna być brana pod uwagę przez laboratorium, zwłaszcza w przypadku badania produktów sypkich, będących mieszaniną składników o różnej gęstości, o skłonności do segregacji (pasze sypkie; żywność, np. musli). Homogeniczność składników wpływa przede wszystkim na niepewność pomiaru a także, chociaż w mniejszym stopniu, na wynik badania. Jednak dla oceny zgodności wyników badań z wymaganiami [4, 19, 20], laboratorium powinno w uzasadnionych przypadkach uwzględnić niepewność pomiaru z niepewnością przygotowania próbki.
Proponowany sposób badania homogeniczności próbki laboratoryjnej oparto na rozdzieleniu składowej zmienności analitycznej (powtarzalność) od składowej zmienności technicznej (homogeniczność) i obliczeniu zmienności analitycznej z rozstępu pomiędzy powtórzeniami wg Raportu Technicznego Nordtest [12]. Zastosowane podejście pozwala ponadto na oszacowanie składowej niepewności związanej z przygotowaniem próbki. Problem homogeniczności próbki laboratoryjnej powinien być uwzględniany na etapie walidacji lub sprawdzania metody, poprzez podział próbki laboratoryjnej na minimum 6 próbek do badań i wykonanie oznaczenia analitu w dwóch powtórzeniach w każdej wydzielonej próbce do badań oraz obliczenie odtwarzalności wewnątrzlaboratoryjnej (precyzja pośrednia) CVh+a, uwzględniającej zmienność przygotowania próbki.
Miarą homogeniczności jest względne odchylenie standardowe (współczynnik zmienności CV, %) wyników badania danego składnika w partii materiału. W przypadku pasz uznaje się za właściwą zmienność składnika w mieszance paszowej w zakresie do 10%. Badanie homogeniczności pasz prowadzone jest w ramach urzędowego nadzoru przez upoważnione laboratoria. Od 2018 r. w ramach urzędowego nadzoru wdrożono instrukcję badania homogeniczności produktów paszowych w której homogeniczność obliczana jest na podstawie współczynnika zmienności technicznej CVh . Sposób i przykłady obliczania homogeniczności (zmienności technicznej) mieszanek paszowych w badaniach urzędowych pasz w zależności od rodzaju matrycy i stopnia rozdrobnienia podano w pracy [22].
Podsumowując, próbki pasz o wysokiej zawartości składników mineralnych (np. MPU, premiksy) należy rozdrabniać w młynie planetarnym (kulowym) lub w moździerzu. Nie należy stosować typowych rozdrabniaczy z uwagi na ryzyko przegrzania próbki i zanieczyszczenia wtórne. Dotychczasowe wyniki badań wykonanych w Krajowym Laboratorium Pasz w Lublinie i ocena wyników w badaniach biegłości z udziałem laboratoriów upoważnionych do badań pasz w ramach urzędowego nadzoru, uzasadniają potrzebę zwrócenia większej uwagi na właściwe wyodrębnienie próbki do badań z próbki laboratoryjnej i jej poprawne rozdrobnienie, co wpłynęło na miarodajność i precyzję badań [6].
Bibliografia
[1] Bielecka G., Korol W., 2018. Wpływ przygotowania próbki na wyniki badań składników analitycznych i dodatków paszowych na przykładzie mieszanki paszowej uzupełniającej. Rozdział w monografii pt. „Bezpieczeństwo i jakość handlowa pasz”, cz. I, Pasze Przemysłowe, 27, 3: 39-42.
[2] EN 16159. Animal feeding stuffs – Determination of selenium by hydride generation atomic absorption spectrometry (HGAAS) after microwave digestion (digestion with 65 % nitric acid and 30 % hydrogen peroxide).
[3] EN 17053:2018. Animal feeding stuffs: Methods of sampling and analysis. Determination of trace elements, heavy metals and other elements in feed by ICP-MS (multi-method)
[4] Eurolab Technical Report No. 1/2017 – Decision rules applied to conformity assessment.
[5] Guide to the Expression of Uncertainty in Measurement (GUM). BIPM, IEC, IFCC, ISO, IUPAC, OIML. International Organization of Standardization, Geneva, Switzerland, 1st Edition 1993.
[6] Horwitz W., Albert R., 2006. The Horwitz Ratio (HorRat). A useful index of method performance with respect to precision. J. AOAC International, 89, 1095-1109
ISO/IEC Guide 98-4:2012. Evaluation of measurement data – The role of measurement uncertainty in conformity assessement.
[7] ISO 13528:2015 Statistical methods for use in proficiency testing by interlaboratory comparisons
[8] Korol W., Matyka S., 1982. Biamperometryczne miareczkowanie chlorków w paszach. Chem. Anal. 27: 323-326
[9] Korol W., Bielecka G., Rubaj J., Walczyński S., 2015. Uncertainty from sample preparation in the laboratory, on the example of various feeds. Accred Qual Assur 20: 61-66 DOI:10.1007/s00769-014-1096-x
[10] Korol W., Rubaj J., Bielecka G., 2015. Porównanie praktycznych podejść szacowania niepewności w badaniach pasz. Rozdział w monografii pt. „Ocena jakości i bezpieczeństwa pasz – Cz. II”, Pasze Przemysłowe, Nr 3/4, 48-52.
[11] Korol W., Rubaj J., Bielecka G., Walczyński S., Reszko-Bogusz J., Dobrowolski R., 2017. Criteria for using proficiency test results for estimation of measurement uncertainty: feed analysis example. Accred Qual Assur 22: 83-89 DOI:10.1007/s00769-017-1252-1
[12] Magnusson B., Näykki T., Hovind H., Kryssel M., 2008. Handbook for Calculation of Measurement Uncertainty in Environmental Laboratories. NORDTEST Report TR 537, Version 3, 2008, str. 41 (wydanie polskie: Biuletyn POLLAB Nr 2/51/2008).
[13] PN-EN ISO 6869:2002 Pasze – Oznaczanie zawartości wapnia, miedzi, żelaza magnezu, manganu, potasu, sodu i cynku. Metoda absorpcyjnej spektrometrii atomowej.
[14] PN-EN ISO/IEC 17025:2018:02. Ogólne wymagania dotyczące kompetencji laboratoriów badawczych i wzorcujących.
[15] PN – EN 15111:2008P Artykuły żywnościowe – Oznaczanie pierwiastków śladowych – Oznaczanie zawartości jodu metodą ICP-MS (spektrometria mas z plazmą wzbudzoną indukcyjnie)
[16] PN EN-ISO 6498:2012. Pasze – Wytyczne do przygotowania próbki.
[17] Rozporządzenie Komisji (UE) nr 691/2013 z dnia 19 lipca 2013 r. zmieniającym rozporządzenie (WE) nr 152/2009 w odniesieniu do metod pobierania próbek i dokonywania analiz.
[18] Rozporządzenie Komisji (WE) nr 152/2009 ustanawiające metody pobierania próbek i dokonywania analiz do celów urzędowej kontroli pasz. Dz. Urz. UE L 54/1 z 26.02.2009.
[19] Rozporządzenie Komisji (UE) Nr 939/2010 w sprawie dopuszczalnych tolerancji. Dz. Urz. UE L 274/4 z 21.10.2010.
[20] Rozporządzenie Komisji (UE) 2017/2279 zmieniające załączniki II, IV, VI, VII i VIII do rozporządzenia PEiR (WE) nr 767/2009 w sprawie wprowadzania na rynek i stosowania pasz. Dz. Urz. UE L 328/3 z 12.12.2017.
[21] Schönherr J., Peterhänsel M., 2008. Zur Untersuchung von Futtermitteln Ausgabe der Analysenspielräume des VDLUFA. Kraftfutter 91 (7-8), 20-27.
[22] Walczyński S., Korol W. 2018. Nowe podejście do oceny homogeniczności produktów paszowych. Rozdział w monografii pt. „Bezpieczeństwo i jakość handlowa pasz”, cz. I, Pasze Przemysłowe, 27, 3: 57-64.
*Waldemar Korol, Grażyna Bielecka, Jolanta Rubaj, Sławomir Walczyński
Instytut Zootechniki-Państwowy Instytut Badawczy
Dział Analityki laboratoryjnej, Krajowe Laboratorium Pasz w Lublinie