Zastosowanie metod spektroskopowych w analizie QSAR
Autor: Monika Kalinowska*
W ostatnich latach bardzo silnie zaznacza się – w badaniach naukowych – trend przewidywania z parametrów strukturalnych związku chemicznego jego właściwości biologicznych i/lub fizykochemicznych. Analiza taka nosi nazwę QSAR (ang. Quantitatiwe Structure – Activity Relationship) czyli badania ilościowej zależności między strukturą chemiczną związku a jego właściwościami biologicznymi/fizykochemicznymi.
Wynikiem analizy QSAR jest równanie, które może być użyte, np. do oceny aktywności biologicznej hipotetycznych związków przy założeniu, że mechanizm ich działania jest taki sam. Takie podejście do badań jest bardzo korzystne gdyż umożliwia: (1) ograniczenie ilości syntetyzowanych i poddawanych wstępnej ocenie związków, (2) projektowanie związków chemicznych o najkorzystniejszych parametrach biologicznych lub fizykochemicznych. Aby otrzymać równania QSAR należy: (1) aktywność biologiczną wyrazić ilościowo, np. jako minimalną ilość (stężenie) pewnej substancji wywołującą określoną odpowiedź biologiczną oraz (2) parametry fizykochemiczne charakteryzujące ligandy wyrazić w postaci liczbowej. W ten sposób otrzymujemy tzw. deskryptory, które opisują różne właściwości cząsteczek, np.: (1) deskryptory elektronowe: moment dipolowy, polaryzowalność, energia HOMO i LUMO, ładunek całkowity, stała Hammeta, efekt rezonansowy i indukcyjny, (2) termodynamiczne: logP, refraktywność molowa, (3) strukturalne: pole powierzchni, objętość, liczba wiązań rotujących, parametr Tafta (steryczny), hydrofobowość podstawnika, wiązania wodorowe (donor-akceptor) oraz wiele innych deskryptorów. Rzadko do opisu struktury związku używa się danych spektroskopowych, np. liczb falowych z widm w podczerwieni lub Ramana. Wynika to z bardzo skomplikowanego obrazu struktury związku przedstawionej w postaci widma z często kilkudziesięcioma pasmami o różnych liczbach falowych. Chcąc zastosować w badaniach QSAR dane z widm należy: (1) wybrać do analizy grupę pojedynczych pasm przypisanych wcześniej określonym drganiom cząsteczki (co wiąże się z czasochłonnym etapem przypisywania pasm) lub (2) zastosować analizę statystyczną (np. składowych głównych) celem uzyskania pojedynczych parametrów pozwalających porównać widma badanych związków chemicznych lub zastosować deskryptory spektroskopowe jak np. EVA (ominięty etap przypisywania pasm). W pracy przedstawiono szereg badań, w których podejmowano próby zastosowania danych spektroskopowych do analizy QSAR. Rezultaty tych badań dały częściową odpowiedź na pytania: (1) czy użycie danych spektralnych jest wystarczające do opisu struktury molekularnej związków, (2) czy możliwe jest stworzenie odpowiednich modeli statystycznych wykorzystujących dane spektroskopowe, które określają zależności między strukturą związków a ich aktywnością biologiczną.
Wprowadzenie
Metody badania ilościowej zależności między strukturą chemiczną związków a ich właściwościami biologicznymi (QSAR) polegają na znalezieniu odpowiednich parametrów opisujących właściwości fizykochemiczne cząsteczki i określeniu za ich pomocą przewidywanej aktywności biologicznej tej cząsteczki [1]. Prawdopodobnie pierwszej próby powiązania aktywności związku z jego budową dokonali Grum-Brown i Faser w 1868 roku zakładając, że aktywność biologiczna związku (Φ) jest funkcją jego budowy chemicznej (c):
Φ = f(c)
Od tego ogólnego wyrażenia do rozwoju ilościowej analizy zależności między strukturą a aktywnością związków była długa droga ponieważ niezbędne było zdefiniowanie odpowiednich pomiarów określających aktywność biologiczną „Φ”, oraz właściwych funkcji matematycznych „f” i metod służących do ilościowego opisu struktury związku „c” [2]. Nowoczesne metody analizy QSAR rozpoczęli w 1964 roku Hansch i Fujita [3] oraz Free i Wilson [4]. Obie metody, metoda Hansch’a i metoda Free-Wilson’a, używają analizy regresji wielokrotnej, ale różnią się opisem właściwości chemicznych. Dzisiaj oprócz analizy regresji wielokrotnej używa się wielu innych metod do opisu funkcji „f” w równaniu wyżej. Są to np. analiza składowych głównych, metoda cząstkowych najmniejszych kwadratów (PLS), analiza dyskryminacyjna i inne. Do opisu struktury chemicznej związku używa się również różnych deskryptorów [5]. Można je sklasyfikować jako: (1) wielkości eksperymentalne, takie jak logPow (współczynnik podziału w układzie oktanol/woda), pKa/pKb, dane spektroskopowe, (2) stałe podstawnikowe (elektryczna, hydrofobowa, steryczna), (3) parametry pochodzące z obliczeń kwantowo-mechanicznych i modelowania molekularnego. Natomiast typowym parametrem opisującym aktywność biologiczną są: stężenie molowe związku powodujące określony efekt (np. LC50), stała wiązania, stała asocjacji, stała inhibitora.
Mimo, że analiza QSAR stanowi stosunkowo młodą dziedzinę nauki, w ostatnich latach nastąpił jej gwałtowny rozwój. Wynika to z faktu, iż metody te znalazły powszechne zastosowanie m.in. w przemysłach farmaceutycznym i kosmetycznym jako podstawowe źródło informacji umożliwiających odkrycie nowych substancji o pożądanych właściwościach biologicznych (leków, konserwantów, dezynfekantów). W trakcie analizy QSAR wyznaczane są właściwości fizykochemiczne związków i ocena korelacji z ich aktywnością biologiczną. W dalszej kolejności następuje uszeregowanie związków podobnych strukturalnie z uwzględnieniem ich specyficznych aktywności. Taki sposób uporządkowania pozwala na łatwe wyznaczenie wspólnych fragmentów struktury przestrzennej, które odpowiedzialne są za aktywność biologiczną lub bezpośrednio na nią wpływają. Stosując metody statystyczne można ustalić równanie wiążące określone właściwości fizykochemiczne z aktywnością związku. Równania QSAR pozwalają nie tylko na ocenę biologicznych właściwości nowych związków, ale również są pomocne przy podjęciu decyzji o konieczności ich syntezy i/lub przeprowadzeniu testów biologicznych. Dzięki temu analiza QSAR zmniejsza ilość niepotrzebnych syntez i badań oraz pozwala na odkrycie nowych właściwości związków, które nie pasują do wyznaczonego równaniem trendu.
Przykłady zastosowań analizy QSAR można byłoby długo mnożyć co potwierdziłoby, że jest to technika powszechnie i z dużym sukcesem stosowana w trakcie poszukiwania związków o określonych właściwościach biologicznych [6]. Na przykład, Seward i współp. bazując na współczynnikach podziału lopPow opracowali równania pozwalające przewidzieć toksyczne właściwości mono- i dikarboksylowych alifatycznych kwasów oraz ich soli sodowych w stosunku do Tetrahymena Pyriformis [7]. Smith i współp. stworzyli model pozwalający przewidzieć toksyczność związków fenolowych obecnych w dymie papierosowym [8]. Metody QSAR powszechnie stosowane są podczas projektowania leków przeciwnowotworowych [9]. Watkins i współp. badając czternaście pochodnych kwasu cynamonowego o potencjalnych właściwościach odstraszających ptaki stworzyli matematyczny model oparty na zależnościach między strukturą związku a jego aktywnością biologiczną [10]. Wyniki tych prac mogą pozwolić na bardziej wydajne i ekonomiczne poszukiwania nowych związków odstraszających ptaki i mających zastosowanie w rolnictwie, sadownictwie i ogrodnictwie. W uzyskanym przez nich równaniu struktura związku opisywana została czterema parametrami zależnymi od zmian w rozkładzie ładunku elektronowego cząsteczki, które zostały obliczone przy użyciu metod kwantowo-mechanicznych. Narasimhana i współp. stosując analizę QSAR badali przeciwdrobnoustrojowe właściwości pochodnych kwasu cynamonowego [11]. Wykorzystali teoretyczne dane fizykochemiczne do opisu struktury związków. Sova i współ. opracowali model pozwalający nakreślić drogę syntezy inhibitorów dehydrogenazy 17β-hydroksysteroidowej należących do pochodnych estrów kwasu cynamonowego [12]. Sunnerheim i współp. badali estry kwasów benzoesowych i cynamonowych jako potencjalnych insektycydów stosowanych w zwalczaniu szeliniaka sosnowego (Hylobius abietis) [13]. Zastosowanie analizy QSAR pozwoliło wyodrębnić cechy związków jakimi powinny charakteryzować się skuteczne insektycydy. Badania QSAR wykorzystane zostały do opracowania syntezy nowych pochodnych benzenosulfonamidu (BZS) posiadających właściwości przeciwdrobnoustrojowe. Wyniki wskazują, że zwiększenie ładunku na atomach wodoru H1 i H1’ (oznaczenia autorów) oraz jego zmniejszenie na atomach N3 i O2 powoduje wzrost aktywności pochodnych BZS w stosunku do Bacillus subtilis oraz opornego na metycilinę Staphylococcus aureus [14].
Zastosowanie metod spektroskopowych w analizie QSAR
Stosunkowo rzadko do opisu struktury związku w badaniach QSAR stosuje się dane spektroskopowe jakimi są liczby falowe pasm z widm w podczerwieni lub ramanowskich. Wynika to przede wszystkim z bardzo skomplikowanego obrazu struktury molekularnej cząsteczki przedstawionego w postaci widma oscylacyjno-rotacyjnego. Chcąc zastosować uzyskane dane spektroskopowe do badań „struktura-aktywność” można: (1) przypisać pasma odpowiednim drganiom cząsteczki, lub (2) zastosować metody statystyczne (np. analizę składowych głównych), lub deskryptory bazujące na wszystkich liczbach falowych (lub z wybranego zakresu np. obszaru daktyloskopowego) celem stworzenia pojedynczych parametrów opisujących strukturę molekularną związku na podstawie liczb falowych z widm (rys. 1). W ten sposób uzyskuje się dwa różne podejścia do badań QSAR z zastosowaniem danych spektroskopowych, tzn. można wykonać korelację danych opisujących właściwości biologiczne związku z liczbami falowymi wybranych pojedynczych pasm z widm lub z deskryptorami, które policzono wykorzystując szeroki zakres liczb falowych. Wynikiem końcowym analizy QSAR jest uzyskanie ilościowych informacji o zależnościach między strukturą związku a jego aktywnością biologiczną w postaci równań matematycznych. Aby sprawdzić czy uzyskany model pozwala przewidzieć właściwości biologiczne substancji należy sprawdzić jego skuteczność wykorzystując dane związków o znanych i nieznanych właściwościach biologicznych oraz porównać uzyskane wyniki z tymi, kiedy do badań QSAR zastosowano inne popularne deskryptory (np. logPow).
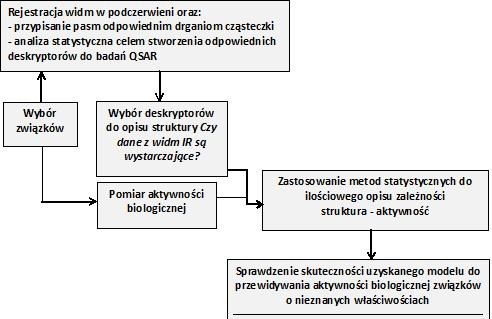
Rys. 1. Schemat analizy QSAR z wykorzystaniem danych spektroskopowych (rysunek autorski)
Przykładem zastosowań do badań „struktura-aktywność” pojedynczych pasm z widm w podczerwieni są prace Cui i współ. [15]. Badali oni działanie szeregu związków zawierających grupę nitrozylową NO (np. Na2[Fe(CN)5NO], K3[Cr9CN)5NO] i inne) na bakterie Clostridium sporogenes. Ustalili, że jedynie grupa NO jest odpowiedzialna za działanie bakteriostatyczne związku, podczas gdy pozostała część cząsteczki nie ma wpływu na rozwój badanych mikroorganizmów. Jednocześnie powiązali właściwości przeciwdrobnoustrojowe proponowanych związków z liczbą falową pasma pochodzącego od drgania rozciągającego N-O w widmie w podczerwieni. Zaproponowali wykorzystanie liczby falowej tego pasma jako „wskaźnika” aktywności przeciwdrobnoustrojowej związków o podobnej budowie chemicznej – wysoka liczba falowa pasma drgania rozciągającego N-O może być wstępnym wskaźnikiem dobrych właściwości bakteriostatycznych związku zawierającego taką grupę. Pewne wątpliwości może budzić fakt całkowitego zaniedbania przez autorów wpływu pozostałej części cząsteczki na rozkład ładunku elektronowego w grupie N-O, a więc również na liczbę falową drgania pasma „wskaźnikowego”. Z drugiej strony, istnieje szereg publikacji, w których przeprowadzona analiza statystyczna wykazała istnienie istotnych korelacji między pojedynczymi wybranymi pasmami z widm w podczerwieni związków chemicznych a parametrami strukturalnymi takimi jak stałe podstawnikowe Hammeta [16] czy parametrem Tafta [17] lub parametrami jonów metali (w przypadku związków kompleksowych) [18]. Oznacza to, że wybór pojedynczych pasm z widm IR do analizy „struktura-aktywność” w pewnych przypadkach może być uzasadniony, ale musi być poprzedzony czasochłonnym procesem przypisywania pasm z widm odpowiednim drganiom cząsteczki. Pinovka w swoich pracach również wykorzystał pojedyncze pasma z widm w podczerwieni i ramanowskich do badania zależności między strukturą wybranych izoflawonów, kumaryn i benzoksazoli a ich właściwościami receptorowymi (względem receptora ER-β) [19]. Udało mu się zidentyfikować pasma z widm badanych związków, które korelują z parametrami określającymi siłę wiązania z receptorem. Wyniki analizy statystycznej wskazywały, że siła wiązania receptora wyraźnie zależy od gęstości elektronowej układu wiązań π.
Zastosowanie w analizie QSAR deskryptorów, które opierają się na większej liczbie pasm z widm (np. z zakresu obszaru daktyloskopowego), pozwala pominąć etap analizy widm i przypisywania pasm. Pierwszy tego typu spektroskopowy deskryptor bazujący na liczbach falowych z widm IR lub Ramana zaproponowali Turner i współp., tzw. deskryptor EVA, który z dobrym rezultatem zastosowali w badaniach specyficznych właściwości steroidów [20-22]:
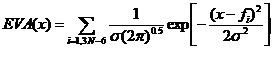
Poszczególne symbole oznaczają: fi – liczba falowa i-tego drgania normalnego, σ – współczynnik krzywej Gaussa opisującej kształt pasma (stały dla całego widma).
Pierwszym etapem badań jest uzyskanie obliczeniowych widm w podczerwieni (lub ramanowskich) najczęściej w zakresie 0-4000 cm-1. Następnie dla każdego pasma o określonej liczbie falowej liczy się deskryptor EVA na podstawie powyższego wzoru. Obliczenia wykonuje się dla każdej liczby falowej z widm wybranych związków, a następnie tworzy się macierz gdzie rzędami są wybrane do badań związki a kolumnami policzone deskryptory EVA w ilościach 4000/L (gdzie L to tzw. „rozdzielczość”, zakres liczb falowych dla których liczone są deskryptory EVA). Dane takie poddawane są analizie statystycznej, najczęściej wielokrotnej regresji liniowej z użyciem metody najmniejszych kwadratów (PLS, ang. Partial Least Squares). Zwykle liczba pasm obecnych w widmach jest inna dla różnych związków, z wyjątkiem tych o jednakowej liczbie atomów (liczba drgań normalnych dla nieliniowej cząsteczki N-atomowej jest równa 3N-6, a dla cząsteczki liniowej 3N-5). Dlatego trudno jest ocenić, które pasma powinny być wybrane i porównywane między sobą jeśli mamy do czynienia z widmami kilku lub większej ilości związków. Użycie deskryptoru EVA pozwala wykorzystać w analizie QSAR jednakową dla wszystkich analizowanych związków liczbę parametrów (w ilości 4000/L) obliczonych na podstawie ustalonych zakresów liczb falowych (L). Liczne badania wykazały, że EVA może być z dużym powodzeniem użyta w analizie QSAR różnych strukturalnie klas związków chemicznych posiadających odmienne właściwości biologiczne [23, 24].
Badania nad wykorzystaniem informacji jakie niosą widma w podczerwieni w analizie QSAR badali również Benigni i współp. Przeprowadzili analizę QSAR na 91 związkach wykorzystując pasma obecne w widmach FT-IR w zakresie 1500-600 cm-1 rejestrowane dla prób w stanie gazowym [25]. Stosując analizę składowych głównych porównywali uzyskane wyniki analizy QSAR z osiągniętymi wcześniej gdy do badań wykorzystane zostały inne niż spektroskopowe deskryptory opisujące strukturę związku. Tylko w jednym przypadku uzyskano lepsze korelacje między spektroskopowymi deskryptorami a aktywnością biologiczną, niż miało to miejsce w przypadku korelacji z pozostałymi deskryptorami. Według autorów widma IR nie mogą być jedynym źródłem opisu struktury związku w analizie QSAR, a do uzyskania dobrych modeli QSAR powinny być stosowane takie strukturalne deskryptory, które w większym stopniu odzwierciedlają mechanizm biologicznego działania związków. Poprzednie badania przeprowadzone przez tych samych autorów na grupie 121 różnych strukturalnie związków wykazały brak wyraźnych korelacji pomiędzy danymi z eksperymentalnych widm w podczerwieni a innymi zwykle stosowanymi deskryptorami (takimi jak logPow, refraktywność molowa, energia orbitalu HOMO, logPow, polaryzowalność). Według autorów informacje o strukturze związku jakie odzwierciedla widmo w podczerwieni są odmienne niż opisują to wybrane do badań deskryptory strukturalne [26]. Benigni i współp. badali również zastosowanie danych z widm w podczerwieni do podziału wybranych związków chemicznych na grupy, które wykazują różny mechanizm działania toksycznego w środowisku wodnym. Liczby falowe z widm w podczerwieni poddawane były analizie składowych głównych, następnie uwzględniano 10 pierwszych składowych w analizie statystycznej z danymi biologicznymi opisującymi toksyczność związków. I tym razem dane spektroskopowe okazały się niewystarczającym parametrem do podziału związków na grupy o różnej aktywności biologicznej.
Wnioski
Przedstawione wyniki badań wskazują, że w niektórych przypadkach udało się z dużym sukcesem zastosować spektroskopowe deskryptory w analizie QSAR (liczby falowe pojedynczych pasm lub policzone deskryptory np. na podstawie analizy składowych głównych). Szczególnie dobrze dopasowane modele QSAR otrzymano stosując deskryptor EVA. Pozwala on porównać poszczególne zakresy widm różnych związków (określone parametrem L) bez konieczności przypisywania pasm drganiom cząsteczki. Wykazano również wysoką korelację tego deskryptoru z innymi popularnie stosowanymi w analizie QSAR parametrami strukturalnymi. Jednak brak wyraźnych korelacji między parametrami spektroskopowymi a danymi biologicznymi, które zostały opisane w innych pracach, nasuwa pytanie: w jakich przypadkach lepsze do analizy QSAR są deskryptory oparte na danych spektroskopowych a kiedy należy stosować inne desktyptory? Prawdopodobnie wtedy możliwe jest zastosowanie do badań QSAR widm IR i Ramana jeżeli zmiany we właściwościach biologicznych różnych związków odzwierciedlane są zmianą położenia pewnych pasm w widmach. Ponieważ widma w podczerwieni stanowią unikalny zapis struktury związku (szczególnie dotyczy to daktyloskopowego zakresu widma) stanowią znakomite źródło danych analitycznych do analizy QSAR. Z tego też powodu budzą ciągłe zainteresowanie wielu ośrodków naukowych zajmujących się poszukiwaniem odpowiednich deskryptorów do opisu struktury związku w badaniach QSAR. Problem stanowi odpowiednie przygotowanie danych z widm w podczerwieni aby mogły mieć one zastosowanie w badaniach ilościowych zależności między strukturą związków a ich aktywnością biologiczną.
Literatura
1. P. Kafarski, B. Lejczak, Warszawa, 1994, PWN.
2. R. Benigni, “Quantitative structure – activity relationship (QSAR) models of mutagens and carcinogens”, CRC Press LLC, U.S.A., 2003.
3. C. Hansch, T. Fujita, J. Am. Chem. Soc. 86 (1864) 1616.
4. S.M. Free Jr., J.W. Wilson, J. Med. Chem. 7 (1964) 395
5. R. Todeschini, V. Consonni, “Handbook of molecular descriptors”, Wiley-VCH, Weinheim, 2000.
6. T. Puzyn, J. Leszczyński, M.T.D. Cronin, “Recent advances in QSAR studies. Methods and applications”, Springer Science+Business Media B.V. 2010.
7. J.R. Seward, T.W. Schultz, SAR and QSAR in Environ. Res. 10 (1999) 557.
8. C.J. Smith, T.A. Perfetti, M.J. Morton, A. Rodgman, R. Garg, C.D. Selassie, C. Hansch, Toxicol. Sci. 69 (2002) 265.
9. V. Markovic, S. Eric, Z.D. Juranic, T. Stanojkovic, L. Joksovic, B. Rankovic, M. Kosanic, M.D. Joksovic, Bioorg. Chem. 39 (2011) 18.
10. R.W. Watkins, J.A. Lumley, E.L. Gill, J.D. Bishop, S.D. Langton, A.D. MacNicoll, N.R. Price, M.G.B. Drew, J. Chem. Ecol. 25 (1999) 2825.
11. B. Narasimhana, D. Belsareb, D. Pharandeb, V. Mouryac, A. Dhakea, Eur. J. Med. Chem. 39 (2004) 827.
12. M. Sova, A. Perdih, M. Kotnik, K. Kristan, T.L. Rižner, T. Solmajer, S. Gobec, Bioorg. & Med. Chem. 14 (2006) 7404.
13. K. Sunnerheim, A. Nordqvist, G. Nordlander, A.-K. Borg-Karlson, C.R. Unelius, B. Bohman, H. Nordenhem, C. Hellqvist, A. Karlén, J. Agricul. Food Chem. 55 (2007) 9365.
14. M.M. González-Chávez, F. Méndez, R. Martínez, C. Pérez-González, F. Martínez-Gutiérre, Molecules 16 (2011) 175.
15. X. Cui, C. Joannou, M. N. Hughes, R. Cammack, FEMS Microbiol. Lett. 98 (1992) 67.
16. M. Liler, Chem. Commun. (London) (1965) 244.
17. R. T. C. Brownlee, R. D. Topsom, Spectrochim. Acta Part A 31 (1975) 1677.
18. W. Lewandowski, M. Kalinowska, H. Lewandowska, J. Inorg. Biochem. 99 (2005) 1407.
19. D. Pinovka, Appl. Spectrosc. 58 (2004) 323.
20. D.B. Turner, P. Willett, A.M. Ferguson, T. Heritage, J. Comp.-Aided Mol. Des. 11 (1997) 409.
21. D.B. Turner, P. Willett, Eur. J. Med. Chem. 35 (2000) 367.
22. D.B. Turner, P. Willet, J. Comp.-Aided Mol. Des. 14 (2000) 1.
23. D.B. Turner, P. Willett, A.M. Ferguson, T.W. Heritage, J. Comp.-Aided Mol. Des. 13 (2000) 271.
24. O. Oltulu, M.M. Yaşar, E. Eroğlu, Eur. J. Med. Chem. 44 (2009) 3439.
25. R. Benigni, A. Giuliani, L. Passerini, J. Chem. Inform. Comp. Sci. 41 (2001) 727.
26. R. Benigni, L. Passerini, D.J. Livingstone, M.A. Johnson, A. Giuliani, J. Chem. Inform. Comp. Sci. 39 (1999) 558.
*Monika Kalinowska, Politechnika Białostocka, Wydział Budownictwa i Inżynierii Środowiska, Zakład Chemii, ul. Zamenhofa 29, 15-435 Białystok